
Инструкция по микробиологическому и технохимическому контролю дрожжевого производства
ИНСТРУКЦИЯ ПО МИКРОБИОЛОГИЧЕСКОМУ И ТЕХНОХИМИЧЕСКОМУ КОНТРОЛЮ ДРОЖЖЕВОГО ПРОИЗВОДСТВА
УТВЕРЖДЕНА: Главным инженером Упрхлеба Минпищепрома СССР А.С.Гришиным
Дата утверждения 6 октября 1983 г.
ИЗДАНО по заказу НПО хлебопекарной промышленности
ВВЕДЕНИЕ
Производство хлебопекарных дрожжей - одно из самых трудных по технологии производств, связанных с жизнедеятельностью микроорганизмов.
Сложный и непостоянный состав сырья и вспомогательных материалов, наличие в них вредных примесей, многостадийный, быстро протекающий процесс выращивания дрожжей - все эти и многие другие особенности производства обусловливают особую потребность в хорошо продуманном микробиологическом и технохимическом контроле производства.
За последние годы в промышленности произошли значительные изменения как в технологии, так и в оборудовании. Уменьшилась кратность разбавления водой мелассы, следовательно, увеличилось содержание сухих веществ в питательной среде. Маточные дрожжи выращиваются с меньшим количеством стадий, используются более продуктивные расы дрожжей.
Улучшилась техническая оснащенность предприятий, появились модернизированные дрожжерастильные аппараты с улучшенной системой воздухораспределения и охлаждения. Для прессования, формовки и упаковки прессованных дрожжей установлены автоматические непрерывнодействующие линии. Для прессования, сушки и упаковки сушеных дрожжей разрабатываются и внедряются также непрерывно действующие линии.
Возросли требования к качеству прессованных дрожжей (утвержден новый ГОСТ 171-81). Улучшение качества продукции и повышение экономической эффективности (выхода дрожжей с единицы сырья) не возможны без правильно поставленного микробиологического и технохимического контроля производства.
Контроль производится работниками заводской лаборатории, ОТК и работниками производства, выполняющими технологические операции непосредственно у рабочего места. Данные контроля должны не только отражать работу завода, но и помогать технологам руководить процессом.
В последние годы разработаны новые методы контроля сырья и вспомогательных материалов, а также методы контроля процесса выращивания дрожжей. Усовершенствованы и доработаны некоторые ранее применявшиеся методы.
В связи с этим в Инструкции изложены как известные, так и вновь разработанные сотрудниками ВНИИХПа и его Ленинградского и Харьковского отделений методы контроля.
Изложенные в Инструкции экспрессные методы позволят вовремя сигнализировать о нарушениях технологических режимов и дадут возможность их нормализовать.
Схема лабораторного и производственного контроля, приведенная в Инструкции, содержит перечень объектов контроля, контролируемых показателей и периодичность их определения.
Микробиологические и химико-технологические методы контроля изложены в двух отдельных разделах для большего удобства пользования Инструкцией специалистами разного профиля - микробиологами и химиками-технологами.
Настоящая Инструкция подготовлена во ВНИИХПе О.А.Бакушинской, Л.Д.Беловой, В.И.Букановой, А.Ф.Лозенко и Н.М.Семихатовой.
МИКРОБИОЛОГИЧЕСКИЙ КОНТРОЛЬ
АНАЛИЗ МЕЛАССЫ
Мелассу для микробиологического анализа отбирают в стерильную посуду в количестве около 100 г из средней пробы, доставленной в лабораторию завода (см. "Отбор проб").
Степень обсемененности мелассы микроорганизмами определяют по двум показателям: общему количеству микроорганизмов в 1 г мелассы; видовому составу (процентное соотношение) микрофлоры, в частности групп микроорганизмов, являющихся вредными для дрожжевого производства.
Посев мелассы производят одновременно как в глубь агаровых питательных сред, так и на поверхность. Для посева используют следующие питательные среды: агар-дрожжевая вода с содержанием 4% сахарозы для учета общего количества микроорганизмов в 1 г; агар-сусло с мелом для выявления кислотообразующих и спорообразующих бактерий; молочный агар для выявления гнилостных бактерий.
Анализ производят по следующей схеме: подготовка питательных сред, разбавление материала, посев, учет количества микроорганизмов, определение состава микрофлоры.
Все необходимые питательные среды должны быть приготовлены заранее и простерилизованы (см. приложение II).
Для анализа одного образца мелассы используют следующие среды: четыре пробирки агар-дрожжевой воды с содержанием 4% сахарозы (количество среды в каждой пробирке должно быть не менее 15 мл), две - с агар-суслом, две - с водяным агаром. Все отобранные пробирки помещают в водяную баню, нагревают до кипения. Когда агар расплавится и станет жидким, без комков, баню снимают с нагревательного прибора и оставляют для охлаждения. Как только температура воды в бане снизится до 65-70 °С, вынимают пробирку с агаром-суслом и всыпают в нее 0,3-0,4 г стерилизованного измельченного мела при соблюдении всех правил асептики. Смесь тщательно перемешивают и выливают в стерильную чашку Петри. Среду равномерно распределяют по дну, следя за тем, чтобы мел не осел на дно.
Подготовка молочного агара заключается в следующем. В расплавленный водяной агар добавляют стерильное обезжиренное молоко (30-40%), перемешивают и оставляют в водяной бане вместе с остальными пробирками. Пробирки охлаждают в водяной бане до температуры 48-50 °С.
Подготовка исследуемого образца для анализа - разведение. Мелассу в количестве 20 г отвешивают на технохимических весах в стерильном химическом стакане (вместимостью 250 мл) и смешивают ее с 180 мл стерильной водопроводной воды - первое разведение (в 10 раз). После тщательного размешивания из первого разведения стерильной градуированной пипеткой отбирают 1 мл и добавляют 9 мл стерильной водопроводной воды - второе разведение (в 100 раз).
Определение общего количества микроорганизмов в 1 г
По 1 мл мелассового раствора из второго разведения вносят в две стерильные чашки Петри (на дно) и заливают расплавленным агаром с дрожжевой водой и сахарозой (4%). Температура агаровой среды должна быть не выше 48-50 °С. Внесенный материал осторожно, круговым движением, перемешивают с питательной средой и дают агару застыть - посев внутрь среды.
Чашки с посевами ставят в термостат при температуре 30 °С. Через 48-72 ч подсчитывают все выросшие колонии и определяют количество микроорганизмов в 1 г мелассы.
Пример. При посеве из второго разведения (в 100 раз) в чашке выросло 45 колоний, следовательно, в 1 г мелассы содержится 45·100=4500 микроорганизмов.
Определение состава микрофлоры мелассы
Поверхностный посев на агар-сусло с мелом для выявления кислотообразующих и агглютинирующих бактерий. Две капли раствора мелассы из первого разведения (в 10 раз) наносят стерильной пипеткой на поверхность приготовленной ранее среды агар-сусло с мелом (способ подготовки см. выше) и распределяют их равномерно стерильным шпателем Дригальского. Чашку с посевом ставят в термостат при температуре 30 °С на 48-72 ч. По истечении этого времени на поверхности среды вырастают колонии, образующие зону просветления мела вокруг себя (кислотообразующие и агглютинирующие бактерии), а также колонии без зоны (спорообразующие аэробные бактерии, кокки и др.).
Посев внутрь среды в молочный агар для выявления гнилостной микрофлоры. 1 мл из второго разведения (1x100) вносят на дно стерильной чашки Петри, вливают заготовленную вышеописанным способом молочную среду, перемешивают, дают застыть и помещают в термостат на 48-72 ч. Гнилостные бактерии, как спороносные, так и не образующие споры, в результате протеолиза молочного белка образуют вокруг своих колоний зону просветления.
Основные группы микроорганизмов, встречающиеся в мелассе
Гнилостные бактерии. Это бактерии, образующие и не образующие споры, вырастают при посевах мелассы как на агар-дрожжевой воде с содержанием 4% сахарозы, так и на агаре-сусле с мелом и на молочном агаре.
Колонии этих бактерий разные - сухие, морщинистые или слизистые, иногда с пузырьками газа. На среде агар-сусло с мелом зон просветления не образуют, на молочном агаре образуют - вокруг колоний.
Под микроскопом палочки подвижные или неподвижные, при окрашивании или ясно видны споры, или не обнаруживаются вовсе.
Спорообразующие нитритообразующие бактерии.
В группе спорообразующих бактерий часто встречаются бактерии, восстанавливающие нитраты в нитриты. Для установления этой способности материал из колонии спороносных бактерий петлей переносят в пробирку со специальной средой (дрожжевая вода с содержанием 4% сахарозы и 0,1% KNO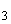 ).
).
Пробирку с посевом ставят в термостат при температуре 30-32 °С. Через 18-24 ч производят качественную пробу на нитриты, для чего 0,5 мл бактериальной культуры смешивают с 0,5 мл реактива Грисса, предварительно нагретого до 90 °С.
Красное окрашивание покажет наличие нитритов в культуральной жидкости, интенсивность же окраски укажет на большую или меньшую способность бактерий восстанавливать нитраты в нитриты.
Дрожжи. Их выявляют в посевах на агар-дрожжевую воду с содержанием 4% сахарозы и на агар-сусло с мелом. Колонии дрожжей на поверхности среды белые или сероватые, чаще с ровным краем, блестящие, многие матовые, морщинистые; в глубине среды - чечевицеобразные, часто с крестообразными выростами, разрывающими агар. Под микроскопом - крупные, овальные или продолговатые клетки с почками или цепочки клеток.
В мелассе встречаются дрожжи различных родов и видов.
Кислотообразующие бактерии. При посеве на агар-сусло с мелом вырастают колонии разной величины (диаметром 0,5-1,5 мм), слегка опалесцирующие, иногда полупрозрачные, с ровными краями, иногда с гладкой поверхностью. Вокруг каждой колонии образуется зона просветления мела диаметром 0,5-2 мм (рис.1).
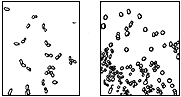
Рис.1. Кислотообразующие бактерии мелассы: лейконосток мезентериодидес (Leuconostok mesenteridides):
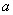 - форма клеток (контрастная окраска с опаль-блау);
- форма клеток (контрастная окраска с опаль-блау); 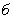 - колонии на сусле-агаре с мелом
- колонии на сусле-агаре с мелом
На агаризованной дрожжевой воде с 4% сахарозы эти же бактерии образуют слизистые бесцветные колонии, в глубине среды чечевицеобразные. Под микроскопом - это довольно мелкие (1,3-1,5 мкм) диплококки и цепочки - стрептококки, по Граму окрашиваются положительно.
Агглютинирующие бактерии. При посеве на агар-сусло с мелом вырастают колонии кислотообразующих бактерий, но более мелкие и с более слабой зоной просветления (диаметр 0,4-0,3 мм); под микроскопом видны цепочки клеток - стрептококки, похожие на кислотообразующие бактерии, по Граму окрашиваются положительно.
Чтобы определить, действительно ли это агглютинирующие бактерии, производят следующее ее определение. В 2-3 мл взвеси, приготовленной из 0,1 г прессованных дрожжей и 10 мл водопроводной воды, петлей вносят бактериальный материал, взятый из колонии. Пробирку несколько раз встряхивают. Наличие склеивания дрожжевых клеток в хлопья указывает на то, что это колонии агглютинирующих бактерий.
Другой быстрый способ проверки заключается в том, что на предметное стекло наносят пипеткой каплю взвеси дрожжей, приготовленную вышеизложенным способом, вносят на петле бактериальный материал, взятый из колонии, и смешивают прямо на стекле этой же петлей. Образование хлопьев свидетельствует о том, что это колонии агглютинирующих бактерий.
Более длительный способ проверки заключается в предварительной разводке бактерий из выросшей колонии. Для этого прокаленной петлей с соблюдением всех правил асептики материал из колонии переносят в пробирку с жидким солодовым суслом с дробиной и 2% мела. Засеянную пробирку помещают в термостат при температуре 30 °С и выдерживают ее там 24 ч. Из полученной разводки бактерий отбирают 1 мл и смешивают с 5-10 мл дрожжевой взвеси, приготовленной вышеописанным способом. Образование хлопьев при встряхивании указывает на агглютинирующие свойства разводки.
Плесени. Колонии на поверхности агаровой среды растут быстро, образуются пушистые дернинки разного цвета - серые, зеленоватые, черные с желтым краем, белые. Под микроскопом видны длинные нити с перегородками или без перегородок и большое количество разной формы (круглых, продолговатых, изогнутых) клеточек - колоний или спор. По морфологии конидий и способам их образования определяют различные роды плесневых грибов.
Кокковая микрофлора. Колонии на поверхности питательных сред мелкие, круглые, желтые или беловатые, слегка морщинистые или гладкие.
Методы окраски микроорганизмов
Простая окраска. Материал, взятый петлей из колонии, вносят в каплю воды на предметном стекле, высушивают и фиксируют в пламени горелки. Подготовленный препарат заливают 1-2 каплями краски (метиленовой синью по Леффлеру или фуксином по Пфейферу - способ приготовления см. приложение II), выдерживают в течение 0,5-1 мин и смывают дистиллированной водой.
Окраска по Граму. Препарат готовят так же, как и для простой окраски: высушивают, фиксируют и заливают карболовым раствором генцианвиолета, выдерживают 2 мин при легком подогревании над пламенем горелки, следят, чтобы краска при этом не подсыхала. Затем избыток краски сливают, заливают препарат раствором йода в йодистом калии и выдерживают в течение 1-2 мин. Далее сливают раствор и быстро опускают препарат в 90%-ный этиловый спирт, выдерживают несколько секунд до тех пор, пока не перестанут отходить струйки краски. После этого препарат промывают водой и заливают фуксином, приготовленным по Пфейферу, на 0,5-1 мин, смывают водой, высушивают фильтровальной бумагой и рассматривают с иммерсионной системой. Грамположительные микроорганизмы окрашиваются в темно-фиолетовый цвет, грамотрицательные - в красный.
Вычисление процентного соотношения микроорганизмов
На плотных агаровых средах в чашках Петри вырастают колонии различных микроорганизмов, соотношение которых выражают в процентах к общему количеству микроорганизмов. При этом учитывают только те чашки, в которых выросло не более 200, но и не менее 30 колоний. Например, если при посеве выросло всего 130 колоний, из них 30 - спорообразующие бактерии, то количество этих бактерий по отношению к общему количеству микроорганизмов составит 30%.
Для получения данных для расчета на поверхности агара-сусла с мелом подсчитывают общее количество всех выросших колоний, а затем отдельно количество колоний спорообразующих, кислотообразующих, кокков, дрожжей и определяют процентное содержание их по отношению к общему количеству колоний.
Пример. Выросло 60 колоний спорообразующих бактерий, 50 колоний дрожжей, 40 колоний кислотообразующих (в том числе и агглютинирующих бактерий), 10 колоний кокков. Всего 160 колоний. В процентах к общему количеству это составит: спорообразующих бактерий 38, дрожжей 31, кислотообразующих бактерий 25 и кокков 6.
Содержание гнилостных бактерий в мелассе и процентное отношение их к общему количеству микроорганизмов определяют путем подсчета колоний в зонах просветления на молочном агаре.
Пример. Общее количество колоний в 1 г мелассы 15 тыс., из них гнилостных - 50, что составит 0,33% к общему количеству.
В результате полученных при микробиологическом анализе данных производится оценка качества мелассы и ее пригодности для переработки в производстве. Так, при наличии в 1 г мелассы не более 2 тыс. микроорганизмов она считается хорошей, от 2 до 20 тыс. - посредственной, свыше 20 тыс. - плохой. Однако большое число (около 90%) спорообразующих бактерий даже в хорошей по обсемененности мелассе из-за возможного присутствия нитритообразующих бактерий дает основание считать ее трудной для переработки, а при наличии дрожжей - опасной при производстве маточных и засевных дрожжей. Кроме того, такая меласса плохо хранится.
Биологическая проба на наличие нитритообразующих бактерий
Мелассу, обсемененную микроорганизмами, быстро образующими нитриты из нитратов (которые всегда содержатся в мелассе), трудно перерабатывать в дрожжевом производстве. Нитриты в количестве 0,001% в среде тормозят рост и почкование дрожжей, а в количестве 0,004% почкование прекращается полностью.
Мелассу разбавляют стерильной водопроводной водой в соотношении 1:10 в стерильной пробирке и ставят в термостат при температуре 30 °С. Качественную пробу на наличие нитритов в пробирке производят через 12, 16, 24, 36 и 48 ч.
По скорости образования нитритов в мелассе, разбавленной водой в соотношении 1:10, т.е. по скорости размножения нитритообразующих бактерий, судят о степени пригодности ее для дрожжевого производства. При появлении через 12-16 ч в пробе с реактивом Грисса даже слабо-розового окрашивания следует принять меры, подавляющие рост бактерий, или уничтожить их, применяя высокотемпературный режим при осветлении, а также антисептики.
Качественная проба на наличие нитритов в питательной среде
В чистую сухую пробирку наливают 0,5 мл первого раствора Грисса и 0,5 мл второго раствора Грисса, нагревают на кипящей водяной бане в течение 1-2 мин и прибавляют 0,2-0,3 мл (по каплям) испытуемого мелассового раствора. Покраснение смеси указывает на наличие нитритов в среде. По интенсивности окрашивания судят о количестве нитритов.
|
Цвет раствора |
Количество нитритов |
|
Ярко-алый |
Очень много |
|
Красный |
Много |
|
Красно-розовый |
Довольно много |
|
Слабо-розовый |
Мало |
Можно пользоваться сухим реактивом Грисса, предварительно разведенным дистиллированной водой в соотношении 1:10.
Выявление наличия нитритообразующих бактерий в кислых мелассах
Этот метод разработан Харьковским отделением ВНИИХПа. На дрожжевые заводы довольно часто поступают кислые мелассы, рН которых составляет 5,2-6,0. Известно, что при таком рН тормозится скорость роста нитритообразующих бактерий, для большинства кислых меласс вышеописанные условия биологической пробы не являются оптимальными и реакция с реактивом Грисса носит отрицательный характер, что приводит к неправильной оценке мелассы по наличию нитритообразующих бактерий. Для этого предварительно следует довести рН мелассового раствора до 7,0-7,2 и только потом выдерживать его в термостате при 30 °С.
С этой целью в отдельной пробе мелассы устанавливают количество щелочи (NaOH), необходимое для доведения раствора мелассы до рН 7,0-7,5. Для этого в небольшой стаканчик отвешивают на технохимических весах 1 г мелассы, добавляют 10 мл стерильной водопроводной воды, размешивают стеклянной палочкой и приливают по 0,1 мл 0,1 н. раствора NaOH (каждый раз замеряя рН), пока в растворе мелассы величина рН не достигнет 7,0-7,5. Проверку рН ведут с помощью потенциометра или с индикаторной бумагой "Рифан".
Затем в стерильную пробирку снова отвешивают 1 г мелассы, добавляют 10 мл дистиллированной водопроводной воды и установленное ранее количество 0,1 н., раствора NaOH для доведения рН до 7,0-7,5; пробирку закрывают ватной пробкой и помещают в термостат при температуре 30 °C.
За скоростью образования нитритов следят, как обычно, при помощи реактива Грисса, отбирая пробы спустя 12, 16, 24 ч и более.
Определение количества нитритов, образовавшихся в мелассе
Этот метод разработан Харьковским отделением ВНИИХПа. Методы определения нитритов (KNO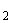 , NaNO) основываются на образовании с нитритами азотных красителей, для чего рекомендуется ряд реактивов. Наиболее распространенным из них является смесь нафтиламина и сульфаниловой кислоты - реактив Грисса, принятый и для качественной реакции определения нитритов.
, NaNO) основываются на образовании с нитритами азотных красителей, для чего рекомендуется ряд реактивов. Наиболее распространенным из них является смесь нафтиламина и сульфаниловой кислоты - реактив Грисса, принятый и для качественной реакции определения нитритов.
Содержание нитритов определяют по интенсивности образующейся при этом розовой окраски сопоставлением оптической плотности испытуемого раствора с оптической плотностью стандартного раствора азотистокислого натрия (NaNO) на фотоэлектроколориметре (ФЭК) или визуально.
Так как собственная окраска мелассы в случае незначительных концентраций нитрита может снизить точность определения, следует предварительно осветлить мелассу. Для этой цели рекомендуется обработка раствора активным углем.
Расчет количества нитритов ведется на содержание NO в граммах или микрограммах (1 г =1000000 мкг) в 100 г мелассы, 1 г NaNO соответствует 0,667 г NO.
Количественное определение накопившихся в пробе мелассы нитритов производят после получения положительной реакции с реактивом Грисса в биологической пробе, принятой для установления нитритообразующих бактерий. Слабо-розовая окраска свидетельствует о том, что в пробе содержится мало нитритов, красная - о том, что много. Оценка мелассы сводится к установлению активности нитритообразования по количеству нитритов, образующихся в биологической пробе.
Методика анализа заключается в следующем. В 2 стерильные конические колбочки вместимостью по 100 мл отвешивают по 5 г мелассы, добавляют в каждую колбочку по 50 мл стерильной водопроводной воды, закрывают ватными пробками, тщательно размешивают и помещают в термостат при температуре 30 °С. Спустя 14-16 ч через каждые 2 ч качественной пробой проверяют появление нитритов, отбирая по 2-3 капли в пробирку с реактивом Грисса.
При оценке интенсивности окраски "мало" 1-ю колбочку берут на количественный анализ, а 2-ю оставляют в термостате.
В отобранную коническую колбочку с мелассой добавляют 2-2,5 г активного угля, тщательно взбалтывают, выдерживают 5-10 мин, количественно переводят содержимое в мерную колбочку вместимостью 100 мл, доводят водой до метки, взбалтывают и фильтруют через складчатый фильтр.
Если спустя 2 ч после взятия на анализ 1-й колбочки интенсивность окраски с реактивом Грисса во 2-й колбочке оценивается уже как "много" нитритов, ее вынимают из термостата и обрабатывают так же, как и 1-ю. При отсутствии интенсивной окраски колбочку с мелассой оставляют еще на 2 ч.
Определение количества нитритов фотоэлектроколориметром (ФЭК). Из 1-й колбочки (качественная оценка "мало") отбирают 5 мл фильтрата (после обработки углем) в мерную колбочку вместимостью 50 мл, добавляют 35 мл дистиллированной воды (до объема 40 мл), 8 мл свежеприготовленного реактива Грисса, доводят объем до метки водой и взбалтывают. Через 15 мин определяют на ФЭК оптическую плотность окрашенного раствора.
Из 2-й колбочки (качественная проба на нитриты "много") отбирают 20 мл фильтрата (после обработки углем) и вносят в мерную колбочку вместимостью 100 мл, доводят дистиллированной водой до метки, взбалтывают и на определение отбирают 5 или 1 мл в мерную колбу на 50 мл, доводят водой до 40 мл (т.е. добавляют воды соответственно 35 или 39 мл), вводят 8 мл реактива Грисса и доводят водой до метки, взбалтывают и спустя 15 мин определяют на ФЭК оптическую плотность окрашенного раствора. Построение калибровочной кривой для этого метода не рекомендовано.
Вместо этого одновременно в трех мерных колбочках на 50 мл готовят стандартный раствор, отбирая в 1-ю колбочку 10, во 2-ю 20 и в 3-ю 30 мл второго стандартного раствора азотнокислого натрия, т.е. по 10, 20, 30 мкг NO. Во все колбочки добавляют воды до объема 40 мл (т.е. 30, 20, 10 мл воды) и по 8 мл реактива Грисса, доводят до метки, взбалтывают и спустя 15 мин устанавливают оптическую плотность стандартных растворов на ФЭК.
Расчет количества нитритов в мелассе ведется следующим образом. В первую очередь устанавливают, какому количеству NO (в мкг) соответствует найденная оптическая плотность раствора мелассы. Например, если оптическая плотность стандартного раствора с 10 мкг NO соответствует 0,09, а найденная оптическая плотность исследуемого раствора мелассы 0,12, то после пересчета это составит 0,12·10/0,09=13,3 мкг NO. Расчет количества нитритов в мелассе в 1-й колбочке (оценка "мало" нитритов) ведется по формуле
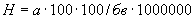 ,
,
где 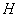 - содержание NO в мелассе, %; - количество NO, найденное в 5 мл фильтрата по оптической плотности исследуемого и стандартного растворов, мкг; - количество осветленного углем фильтрата, взятое для определения оптической плотности раствора, мл;
- содержание NO в мелассе, %; - количество NO, найденное в 5 мл фильтрата по оптической плотности исследуемого и стандартного растворов, мкг; - количество осветленного углем фильтрата, взятое для определения оптической плотности раствора, мл; 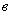 - навеска мелассы, взятая на определение, г; 100 - пересчет на 100 мл фильтрата после осветления мелассы углем; 100 - пересчет на 100 г мелассы; 1000000 - перевод микрограммов в граммы.
- навеска мелассы, взятая на определение, г; 100 - пересчет на 100 мл фильтрата после осветления мелассы углем; 100 - пересчет на 100 г мелассы; 1000000 - перевод микрограммов в граммы.
Пример пересчета. Взято 5 г мелассы и после накопления нитритов, обработки углем и доведения объема до 100 мл на определение отобрано 5 мл фильтрата в мерную колбочку на 50 мл.
Допустим, что оптическая плотность исследуемого раствора 0,124; оптическая плотность стандартного раствора с 20 мкг NO 0,148. Отсюда , т.е. количество NO, найденное в 5 мл фильтрата, составит
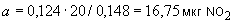 ;
; 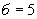 ;
; 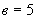 .
.
Общее количество нитритов, накопившихся в пробе мелассы, составит =16,75·100·100/5·5·1000000=/2500=16,75/2500 =0,0067%.
Расчет количества нитритов в 3-й колбочке (оценка "много") ведется по формуле
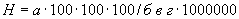 ,
,
где - содержание NO, в мелассе, %; - количество NO, найденное в 5 мл фильтрата по оптической плотности исследуемого и стандартного растворов, мкг; - количество раствора, взятое из 2-й мерной колбочки на определение, мл; - количество осветленного фильтрата мелассы для второго разведения, мл; 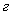 - навеска мелассы, взятая на анализ, г; 100 - пересчет на 100 мл осветленного фильтрата, на 100 мл при втором разведении и на 100 г мелассы; 1000000 - перевод микрограммов в граммы.
- навеска мелассы, взятая на анализ, г; 100 - пересчет на 100 мл осветленного фильтрата, на 100 мл при втором разведении и на 100 г мелассы; 1000000 - перевод микрограммов в граммы.
Пример расчета. Взято 5 г мелассы и после накопления нитритов и обработки углем доведено до 100 мл, 20 мл фильтрата отобрано во 2-ю мерную колбочку и доведено до 100 мл, откуда на определение нитритов отобрано 5 мл в мерную колбочку на 50 мл.
Допустим, что оптическая плотность этого раствора составляет 0,177, оптическая плотность стандартного раствора с 30 мкг NO равна 0,193, откуда: =0,177х30/0,193=27,5 мкг NO; =5; =20; =5.
Общее количество нитритов, накопившееся в пробе мелассы, составляет: 27,5·100·100·100/5·20·5·1000000=/500=27,5/500=0,055% NO.
Если нитритов образовалось "очень много" и на определение из 2-й колбочки взят всего 1 мл, то расчет ведется по формуле 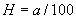 .
.
Определение количества накопившихся в пробе мелассы нитритов визуальным способом.
При определении нитритов в пробе мелассы визуальным способом количество NO устанавливают таким образом. Подбирают ряд пробирок одинакового диаметра. В 4 из них набирают 1, 2, 3, 4 мл второго стандартного раствора NaNO, в 1 мл которого содержится 1 мкг NO. К 1-й пробирке добавляют 3,2 мл, ко 2-й - 2,2, к 3-й - 1,2 и к 4-й 0,2 мл дистиллированной воды так, чтобы в каждой пробирке находилось 4,2 мл.
В две другие пробирки* отбирают 1-2 мл испытуемого осветленного раствора мелассы, добавляют в обе пробирки воды до общего объема 4,2 мл и во все 6 пробирок добавляют по 0,8 мл раствора Грисса до общего объема 5 мл. Содержимое пробирки взбалтывают и спустя 15 мин устанавливают, с какой пробиркой стандартного ряда совпадает окраска испытуемого раствора мелассы.
________________
* В две пробирки испытуемый раствор отбирают для большей точности анализа. При расчете берут среднее трех определений.
При визуальном способе определения нитритов не следует доводить окраску раствора до интенсивного розового цвета, правильнее соответственно уменьшить взятое на анализ количество испытуемой среды.
Расчет количества NO в пробе мелассы ведут следующим образом. Допустим, что при оценке количества нитритов "мало" из колбочки вместимостью 100 мл после осветления мелассы в две пробирки отобрано по 1 мл испытуемого раствора, к которому добавлено 3,2 мл воды и 0,8 мл раствора Грисса. Спустя 15 мин установлено, что окраска этого раствора совпадает с 4-й пробиркой стандартного раствора NaNO, т.е. в ней найдено 4 мкг NO.
Отсюда в 100 г мелассы может накопиться: 4·100·100/1·5·1000000=4/500=0,008% NO, т.е. общая формула расчета составляет =/500%, где а - найденное количество NO во взятом на определение 1 мл раствора мелассы, мкг; 500 - разведение и перевод микрограммов в граммы.
Если в 2 пробирки отобрано не по 1 мл, а, например, по 2 мл испытуемого раствора, а окраска раствора тоже совпала с 4-й пробиркой стандартного раствора, что соответствует 4 мкг NO, тогда в 100 г мелассы содержится 4·100·100/2·5·1000000=4/1000=0,004% NO.
Общая формула расчета в этом случае будет =/1000%.
Если оценка количества нитритов, накопившихся при биологической пробе, характеризовалась как "много", то на определение из 2-й колбочки вместимостью 100 мл после осветления мелассы отбирают всего 0,5 мл в пробирку.
Допустим, что розовая окраска этого раствора совпала с окраской 2-й пробирки стандартного раствора NaNO, что соответствует 3 мкг NO, тогда найденное количество нитритов составит 2·100·100·100/0,5·20·5·1000000=2/50=0,040% NO.
Общая формула расчета при таком разведении будет =/50, где - количество NO в пересчете на 100 г мелассы; - количество NO в 0,5 мл раствора, мкг; 50 - разведение и перевод микрограммов в граммы.
КОНТРОЛЬ ЧИСТОЙ КУЛЬТУРЫ ДРОЖЖЕЙ
Производственную чистую культуру дрожжей накапливают постепенно. Начинают с посева одной петли культуры дрожжей с агаровой питательной среды. Эту петлю вносят в несколько миллилитров жидкой питательной среды.
Через ряд последующих пересевов получают несколько сотен килограммов прессованных маточных дрожжей для дальнейшего производства задаточных и засевных дрожжей.
Процесс размножения чистой культуры дрожжей происходит в несколько стадий: первые стадии - в лаборатории, затем в цехе чистой культуры, в специальных аппаратах, и далее - в производственном цехе, в дрожжерастильной аппаратуре маточного отделения.
После окончания последней производственной стадии дрожжи сепарируют, промывают и прессуют или хранят в виде густой дрожжевой взвеси (дрожжевого концентрата) при температуре 2-4 °С в специальных сборниках; в прессованном виде - в холодильной камере при той же температуре. По мере надобности дрожжи используют при изготовлении задаточных дрожжей (естественной чистой культуры).
От качества культуры дрожжей, применяемой на заводе, зависит в значительной мере качество готовой продукции (прессованных и сушеных хлебопекарных дрожжей).
Чистую культуру любой из производственных рас, применяемых на мелассово-дрожжевых заводах, поддерживают на скошенной агаровой питательной среде, подобранной с таким расчетом, чтобы сохранить активность культуры при размножении ее в условиях дрожжевого производства.
Культуру пересевают не реже чем через 10-12 дней с предварительной подмолодкой в жидком солодовом сусле плотностью 10-12% СВ в течение 16-18 ч при температуре 30 °С. После пересева на скошенный питательный агар (состав среды см. в приложении II) культуру выращивают в течение 24-26 ч и хранят при комнатной температуре.
Сохранение активности культуры и ее производственных качеств является немаловажным фактором для успешного осуществления технологического процесса на дрожжевом заводе.
Оценка качества культуры
Оценка качества культуры производится по трем показателям: чистоте культуры, ферментативной активности генеративной активности. Ниже описаны три взаимодополняющих метода.
Микроскопирование. Контролю подвергают односуточную культуру из жидкого солодового сусла плотностью 10-12% СВ. Одну каплю взвеси с помощью стерильной пипетки наносят на чистое предметное стекло и накрывают покровным стеклом (оба стекла предварительно протирают спиртом). Подготовленный препарат помещают на предметный столик микроскопа и просматривают десять полей зрения при увеличении в 600-800 раз. При просмотре не должно быть обнаружено ни посторонних дрожжевых грибков, ни бактерий.
Посев в чашки Петри. Одну петлю агаровой культуры вносят в пробирку со стерильной водой. После тщательного размешивания 1-2 капли взвеси наносят на поверхность твердой питательной среды (агар-сусло с мелом) и размазывают шпателем Дригальского. Затем этот шпатель переносят во вторую чашку с такой же средой и размазывают остатки на шпателе - посев с переносом или истощающий посев.
Для выявления бактериальной инфекции используется среда с нистатином, методика посева та же. На всех чашках не должно быть обнаружено колоний посторонних микроорганизмов (дрожжей и бактерий).
Посев на элективную (ацетатную) среду. Этим методом выявляются в исследуемом материале даже единичные клетки посторонних дрожжевых грибков.
Одну петлю культуры со скошенного агара вносят в пробирку с ацетатной средой (состав среды см. в приложении II). Посев производят с соблюдением всех правил асептики над пламенем горелки стерильной пипеткой или предварительно прокаленной петлей.
Засеянную пробирку помещают в термостат при температуре 30 °С и ведут за ней ежедневные наблюдения в течение не менее чем 5-6 дней. Появление пленки на поверхности среды или помутнение ее указывают на наличие в культуре посторонних дрожжевых грибов. О степени загрязненности этими микроорганизмами судят по срокам появления пленки или мути. Культура считается чистой, если в течение всего периода наблюдения пленка или муть не появились.
Оценка ферментативной (зимазной и мальтазной) активности культуры
В пробирку с двухсуточной культурой на скошенном питательном агаре наливают 1-2 мл стерильной водопроводной воды и сильно взбалтывают. Полученную густую эмульсию дрожжей (несколько капель) наносят стерильной пипеткой на поверхность мелассовой агаровой среды. Посевной материал тщательно распределяют шпателем Дригальского по поверхности среды. Таким образом засевают не менее двух чашек Петри, которые затем помещают в термостат при 30 °С на 48 ч. Чашки Петри ставят крышкой вниз, чтобы не попал конденсат. Выросшую культуру (налет) дрожжей снимают с поверхности агара химическим шпателем.
Влажность образовавшегося комочка дрожжей близка к влажности прессованных дрожжей, т.е. около 75%. Из полученных таким способом дрожжей берут две навески по 0,5 г каждая для анализа.
Зимазная активность - скорость сбраживания глюкозы или сахарозы; мальтазная активность - скорость сбраживания мальтозы. Как мальтазную, так и зимазную активность выражают во времени, необходимом для выделения 10 мл диоксида углерода при сбраживании 5%-ного раствора мальтозы или глюкозы дрожжами, заданными в количестве 2,5% (в прессованном виде) по отношению к объему сахарного раствора. Величину зимазной и мальтазной активности выражают в минутах.
При определении пользуются микрогазометром системы Елецкого. Микрогазометр состоит из стаканчика и манометрической крышки, в которой имеется изогнутая газоотводная трубка с трехходовым краном, при помощи которого осуществляется соединение газоотводной трубки как с внутренней камерой прибора - стаканчиком, так и с внешней средой (рис.2).
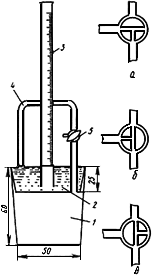
Рис.2. Микрогазометр:
1 - стаканчик; 2 - манометрическая крышка; 3 - измерительная трубка; 4 - газоотводная трубка; 5 - трехходовой кран
Перед началом работы в манометрическую крышку заливают насыщенный раствор поваренной соли, подкрашенный метиленовой синью. Раствор наливают до основания измерительной трубки и этот уровень считают за нуль; все шлифы смазывают вазелином. Измерительную трубку градуируют с точностью до 1 мл.
Высоту 1 мл на измерительной трубке вычисляют по следующей формуле:
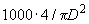 ,
,
где 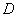 - диаметр измерительной трубки (=8 мм);
- диаметр измерительной трубки (=8 мм); 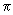 =3,14.
=3,14.
Подставив эти значения в формулу, получим 1000·4/3,14·64=20 мм.
Ход определения заключается в следующем. Отобранные для анализа 0,5 г дрожжей помещают в стаканчик прибора, заливают 10 мл водопроводной воды температурой 35 °С и размешивают их в этой воде. К полученной суспензии дрожжей добавляют 10 мл 10%-ного раствора сахара (сахарозы, глюкозы или мальтозы) и быстро закрывают стаканчик манометрической крышкой, предварительно переведя трехходовой кран в положение , затем в положение для выравнивания давления (см. рис.2) внутри прибора с атмосферным. После этого кран переводят в положение и прибор помещают в термостат при 35 °С, отметив время начала анализа. Наблюдают за ходом брожения и отмечают время, когда солевой раствор в измерительной трубке поднимется до отметки 10 мл, т.е. время, за которое выделилось 10 мл СО.
После окончания анализа кран на газоотводной трубке переводят в положение для того, чтобы жидкость в трубке опустилась, и разъединяют манометрическую трубку и стаканчик. Из стаканчика выливают жидкость, моют и вытирают досуха.
У различных культур, используемых в производстве, зимазная активность колеблется от 40 до 60 мин, мальтазная - от 70-80 до 160 мин.
Определение генеративной активности дрожжей
Показатель генеративной активности дает возможность объективно судить о степени пригодности той или иной культуры дрожжей для производства. Генеративная активность - это способность дрожжей к активному росту и размножению. Описанным ниже методом определяется скорость роста дрожжей в определенных условиях - модуль роста.
Ход определения заключается в следующем. 0,1 г дрожжей в прессованном виде, предварительно выращенных на агаризованной среде, помещают в колбочку вместимостью 300 мл. Добавляют 70 мл мелассы плотностью 8% СВ, осветленной горячим способом, и 0,04 г однозамещенного фосфорнокислого калия, хорошо перемешивают до образования равномерной взвеси и ставят на 6 ч в качалочный аппарат, помещенный в термостат с температурой 30-32 °С.
Количество засевных и выросших дрожжей определяют на приборе ФЭК-56 по оптической плотности или мутности раствора. Перед определением на приборе взвесь дрожжей разбавляют водой. 3 мл взвеси дрожжей помещают в мерную колбу вместимостью 100 мл, доводят до метки водопроводной водой и определяют после тщательного перемешивания оптическую плотность взвеси.
Фотоэлектрический колориметр-нефелометр ФЭК-56 предназначен как для измерения оптической плотности, или светопропускания растворов, так и для измерения светорассеяния взвесей и эмульсий в проходящем свете, т.е., по существу, измеряется тоже оптическая плотность.
В инструкции, приложенной к прибору, описаны принцип действия, устройство прибора (с рисунками и фото), подробно описана методика работы с ним, поэтому эти сведения в данной Инструкции не приведены.
Для определения количества дрожжей во взвеси по оптической плотности пользуются светофильтром N 3, кюветами - 5,0; 6,0 мм (толщина кюветы).
Разбавленную указанным выше способом взвесь дрожжей наливают в одну кювету, а в другую кювету для сравнения наливают раствор мелассы, приготовленный следующим способом: 3 мл раствора мелассы с содержанием 8% СВ помещают в мерную колбу на 100 мл, доводят до метки водопроводной водой и перемешивают.
Оптическую плотность взвеси дрожжей определяют в начале и конце выращивания, полученные значения сравнивают с градуировочной таблицей или кривой и таким образом получают данные о количестве дрожжей во взвеси. Принципы изготовления градуировочных таблиц приведены ниже.
Генеративная активность культуры оценивается по количеству дрожжей, накопившихся за период выращивания (6 ч).
Основным показателем является скорость роста дрожжей - модуль роста. Модуль роста определяют по формуле, предложенной Е.А.Плевако,
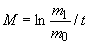 ,
,
где 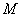 - скорость или модуль роста;
- скорость или модуль роста; 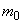 - количество засеянных дрожжей, г;
- количество засеянных дрожжей, г; 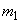 - количество выросших дрожжей, г;
- количество выросших дрожжей, г; 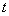 - длительность выращивания, ч.
- длительность выращивания, ч.
У дрожжей с хорошей генеративной активностью модуль роста составляет от 0,20 до 0,23, со средней - соответственно от 0,16 до 0,19, с плохой - от 0,12 до 0,15.
В табл.1 приведена примерная градуировочная таблица для определения концентрации дрожжей во взвеси.
Таблица 1
|
Содержание прессованных дрожжей |
Показания нефелометра |
|
10 |
0,13 |
|
15 |
0,17 |
|
20 |
0,24 |
|
25 |
0,29 |
|
28 |
0,33 |
|
30 |
0,35 |
|
32 |
0,38 |
|
33 |
0,42 |
|
34 |
0,45 |
|
35 |
0,47 |
|
37 |
0,48 |
|
38 |
0,49 |
|
40 |
0,51 |
|
42 |
0,53 |
|
43 |
0,55 |
|
45 |
0,57 |
|
47 |
0,59 |
|
48 |
0,60 |
|
50 |
0,62 |
|
52 |
0,62 |
|
54 |
0,65 |
|
56 |
0,67 |
|
58 |
0,69 |
|
60 |
0,72 |
|
62 |
0,76 |
КОНТРОЛЬ ПРОЦЕССА ПРОИЗВОДСТВА МАТОЧНЫХ ДРОЖЖЕЙ ЧК И ЕЧК
Контроль осуществляется как в лабораторных, так и в производственных стадиях. В конце каждой стадии, перед передачей содержимого аппарата в следующую стадию, отбирают пробы в стерильную посуду в количестве от 2-3 до 300 мл в зависимости от объема жидкости в аппарате.
В каждой пробе производят следующие анализы: определение количества почкующихся клеток, отмерших клеток, посторонних грибков (в %); выявление наличия бактерий.
Все перечисленные выше анализы производят в одной и той же пробе методом микроскопирования. Одну каплю содержимого из аппарата помещают на предметное стекло и накрывают ее покровным стеклом. Приготовленный таким образом препарат помещают на предметный столик микроскопа и просматривают при увеличении в 600-800 раз десять полей зрения.
В процессе микроскопирования определяют: 1) количество почкующихся клеток (в %); для этого в каждом поле зрения считают отдельно почкующиеся и непочкующиеся клетки. Выводят среднее количество тех и других в десяти полях зрения и вычисляют процентное соотношение. Количество почкующихся клеток может значительно колебаться от 10 до 80%; 2) количество отмерших клеток (в %); для этого одну каплю содержимого аппарата смешивают на предметном стекле с 1-2 каплями разбавленной метиленовой сини по Финку (см. приложение II). Количество отмерших клеток во всех стадиях не должно превышать десятых долей процента; 3) наличие посторонних дрожжевых клеток; посторонние дрожжи должны отсутствовать; 4) наличие бактериальной инфекции: во всех десяти полях зрения не должны быть обнаружены как подвижные, так и неподвижные палочки. Единичные неподвижные палочки (диплококки) допускаются в последних производственных стадиях и в прессованных дрожжах ЧК. При наличии посторонних дрожжей и бактериальной инфекции даже не в каждом поле зрения производят более углубленные исследования: посевы на агаризованные среды для выявления характера инфекции (см. следующий раздел).
Оценка степени пригодности маточных дрожжей
Маточные дрожжи получают из чистой культуры той или иной расы дрожжей, выращенной на агаровой среде, путем пересевов и накапливания массы сначала в лаборатории, затем в цехе чистых культур в маточной и производственной аппаратуре. Далее их используют для засева среды дрожжерастильных аппаратов и получения товарных дрожжей.
Маточные дрожжи должны представлять собой технически чистую культуру дрожжей-сахаромицетов, т.е. не должны содержать посторонних дрожжевых грибков и вредных для производства бактерий. Должны обладать высокой мальтазной активностью, стойкостью при хранении и осмоустойчивостью, высокой генеративной активностью и обеспечивать быстрый прирост биомассы при выращивании товарных дрожжей.
Выявление посторонних дрожжей и бактерий
Основным критерием для определения степени пригодности маточных дрожжей для производства товарных должна быть чистота культуры и в первую очередь отсутствие посторонних дрожжей и вредных для производства бактерий.
Определение процентного соотношения основной культуры и посторонних микроорганизмов. Для определения процентного соотношения сахаромицетов и посторонних микроорганизмов (дрожжей и бактерий) применяют метод посева на сусло-агар с мелом. Посев производят над пламенем горелки с соблюдением всех правил асептики в боксе или в отдельной чистой комнате.
1-2 капли среды с дрожжами из аппарата или комочек прессованных дрожжей переносят пастеровской пипеткой или предварительно прокаленной петлей в 10 мл стерильной водопроводной воды, хорошо перемешивают, это первое разведение.
Из первого разведения 1-2 капли переносят во вторую пробирку со стерильной водопроводной водой и опять хорошо перемешивают - второе разведение.
Из второго разведения 2-3 капли взвеси пипеткой наносят на поверхность агаровой среды (сусло-агар с мелом) и распределяют по поверхности среды шпателем Дригальского. Затем шпатель переносят на поверхность питательной среды во вторую чашку Петри и тщательно размазывают по ее поверхности оставшийся на шпателе посевной материал. Такой метод истощающего посева позволяет получить либо на первой, либо на второй чашке количество колоний, удобное для подсчета, т.е. не более 30-40.
Чашки с посевами ставят в термостат при 30 °С крышкой вниз и выдерживают там в течение 48 ч, после чего подсчитывают выросшие колонии дрожжей и бактерий. Подсчитывают отдельно колонии основной культуры (сахаромицетов), посторонних дрожжевых грибков и бактерий и вычисляют процентное соотношение их.
Внешний вид колоний всех этих микроорганизмов различен: сахаромицеты образуют колонии белого или слегка желтоватого цвета, выпуклые, с гладкой блестящей поверхностью и ровным краем; посторонние грибки (несахаромицеты) образуют колонии, различные по внешнему виду в зависимости от рода и вида этих грибков: плоские или слегка выпуклые, сероватого цвета или белые, с поверхностью слегка складчатой или матовой, ворсинчатой, с ровным, фестончатым или ворсинчатым краем; из бактерий чаще всего присутствуют кислотообразующие бактерии (лейконосток, агглютинирующие бактерии), спорообразующие бактерии и кокковые формы (сарцины или педиококки).
Кислотообразующие и агглютинирующие бактерии образуют колонии, по внешнему виду похожие между собой: круглые, полупрозрачные, слегка опалесцирующие, просветляющие мел и образующие различной интенсивности и ширины зону просветления вокруг своих колоний, диаметр колоний от 0,5 до 1 мм.
Спорообразующие бактерии образуют довольно крупные колонии диаметром 3-10 мм, сухие или слизистые, с ровным или фестончатым краем. Педиококки или сарцины образуют желтые, реже белые, складчатые или гладкие колонии разного размера.
Пример расчета для засевных дрожжей. На всей поверхности чашки выросло 49 колоний дрожжей-сахаромицетов, одна колония посторонних дрожжей (всего 50) и две колонии кислотообразующих бактерий (колоний с зоной просветления, всего 52).
Количество посторонних дрожжей определяют следующим образом:
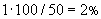 посторонних дрожжей.
посторонних дрожжей.
Количество бактерий в процентах вычисляют по отношению к общему количеству микроорганизмов. Например, в данном случае общее количество составляет 52, отсюда количество кислотообразующих бактерий составит
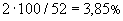 .
.
Таким образом, при содержании посторонних дрожжей выше допустимого такие дрожжи ЧК не могут быть использованы для изготовления ЕЧК, а должны быть использованы только как засевные для товарных аппаратов.
Определение степени зараженности посторонними микроорганизмами содержимого дрожжерастильных аппаратов и дрожжей ЧК и ЕЧК. Этот метод разработан Ленинградским отделением ВНИИХПа для определения абсолютного количества клеток различных групп и видов вредной микрофлоры как в процессе производства, так и в дрожжах ЧК и ЕЧК, т.е. в 1 мл культуральной среды, дрожжевого концентрата или в 1 г дрожжей.
Дрожжевому производству свойственна определенная, характерная для него посторонняя микрофлора, которую условно можно подразделить на следующие физиологические группы: молочнокислые (кислотообразующие и агглютинирующие) бактерии; гнилостные бактерии (большинство спорообразующих и часть не образующих споры); бактерии кишечной группы; посторонние дрожжи.
Для выявления этих групп микроорганизмов используют питательные среды определенного состава, а именно такие, на которых данный вид микробов имеет преимущественное развитие и может быть отличим по виду колоний от других микроорганизмов. Например, для обнаружения всех видов бактерий в питательные среды вносят антибиотик нистатин, обладающий губительным действием на дрожжи (фунгицидным). Этот препарат нужен для того, чтобы на чашках Петри не вырастали дрожжи, а колонии бактерий становились хорошо заметными.
Ниже приведены питательные среды для выявления приведенных выше групп микроорганизмов.
|
Группа микроорганизмов |
Средства для выявления и учета |
|
Молочнокислые бактерии (лактобактерии и лейконосток) |
Сусло-агар с мелом и нистатином |
|
Лейконосток |
Дрожжевой агар с сахарозой и нистатином |
|
Гнилостные бактерии |
Молочный агар с нистатином |
|
Посторонние дрожжи |
Сусло-агар, синтетическая среда с лизином |
Для выявления и количественного учета производится посев исследуемого материала на приведенные выше среды. На дрожжевом агаре с сахарозой и нистатином вырастает большая часть инфицирующих бактерий, поэтому на этой среде подсчитывается общее количество бактерий в пробе в пересчете на единицу объема или массы. На этой же среде отдельно учитывается лейконосток; он образует характерные прозрачные каплевидные колонии.
На молочном агаре с нистатином выявляются гнилостные бактерии, вокруг их колоний образуются прозрачные зоны в результате расщепления белка молока.
Общая численность остальных бактерий может быть определена по разности между общим количеством бактерий, выросших на дрожжевом агаре с сахарозой, и количеством молочнокислых и гнилостных бактерий, выросших соответственно на сусле-агаре с мелом и молочном агаре.
Техника выполнения количественного посева состоит в следующем. Для обеспечения достаточной точности анализа необходимо определенный объем (1-10 мл) или массу (1 г) исследуемого материала развести стерильной водопроводной водой до такой степени, чтобы в 1 мл суспензии содержалось 20-100 клеток посторонних микроорганизмов. При посеве 1 мл такой суспензии в чашку Петри из каждой клетки разовьется колония, и по числу выросших колоний можно подсчитать число клеток в исходном материале.
Так как трудно предугадать, во сколько раз следует развести водой пробу, чтобы в 1 мл суспензии оказалось от 20 до 100 клеток посторонних микроорганизмов, проводят несколько последовательных разведений одной и той же пробы и из них производят посев. После выращивания в термостате при 30 °С отбирают для анализа и подсчета те чашки, где разведение соответствует требуемому, т.е. в чашке вырастает 20-100 (не более 300) колоний. Число выросших колоний умножают на степень разведения пробы и вычисляют количество клеток в 1 г дрожжей (учитывая, что одна клетка образует одну колонию).
Например, на чашке Петри выросло 50 колоний молочнокислых бактерий, посев в эту чашку проведен из разведения дрожжей 1:100 тыс., значит, число молочнокислых бактерий в 1 г дрожжей равно 50·100 тыс. = 5 млн. клеток. Посев, как правило, осуществляется в две параллельные чашки, и для расчета берутся средние данные.
Техника последовательных разведений в стерильном стеклянном стаканчике (с бумажным колпачком) состоит в следующем. Отвешивают 1 г прессованных дрожжей или отбирают 1 мл бродящей жидкости из аппарата. В стаканчик наливают небольшое количество стерильной водопроводной воды из колбы со 100 мл этой воды. Дрожжи тщательно размешивают и растирают стерильной стеклянной палочкой, чтобы получилась однородная суспензия. Всю суспензию дрожжей из стаканчика возвращают в ту же колбу со 100 мл воды. Всю операцию проводят с соблюдением правил асептики (над пламенем горелки). Далее из колбы производят ряд последовательных разведений в стерильной воде, где с каждым последующим переносом разведение суспензии увеличивается в 10 раз. Рекомендуется при каждом новом переносе пользоваться новой, неиспользованной стерильной пипеткой.
При анализах дрожжей ЧК, ЕЧК и содержимого аппаратов рекомендуются следующие схемы посевов для обнаружения и подсчета посторонних микроорганизмов (табл.2).
Таблица 2
|
Микроорганизмы |
Питательная среда |
Разведение |
||||||
|
100 |
1000 |
10 тыс. |
100 тыс. |
1 млн. |
10 млн. |
100 млн. |
||
|
Дрожжи культурные и посторонние |
Сусло-агар |
- |
- |
- |
- |
- |
2 чашки |
2 чашки |
|
Посторонние дрожжи |
Синтетическая среда с лизином |
- |
2 чашки |
2 чашки |
2 чашки |
- |
- |
- |
|
Бактерии и лейконосток |
Дрожжевой агар с сахарозой и нистатином |
- |
- |
- |
2 чашки |
2 чашки |
2 чашки |
- |
|
Молочнокислые бактерии (лактобактерии с лейконостоком) |
Сусло-агар с мелом и нистатином |
- |
- |
- |
2 чашки |
2 чашки |
2 чашки |
- |
|
Гнилостные бактерии |
Молочный агар с нистатином |
- |
2 чашки |
2 чашки |
2 чашки |
- |
- |
- |
После разведения проб производят посев в чашки Петри на указанные агаризованные среды. В стерильную чашку Петри вносят 1 мл суспензии из соответствующего разведения, указанного в табл.2. Далее в чашки, предназначенные для выявления бактерий (сусло-агар с мелом, дрожжевой агар, молочный агар), отдельной стерильной пипеткой со слегка отбитым носиком вносят водную суспензию нистатина. Затем в пробирки с расплавленным суслом-агаром, предназначенные для выявления молочнокислых бактерий, вносят стерильный порошок мела в количестве 0,2-0,3 г (стерилизуют отдельно в пробирках при 170 °С 1 ч). Тщательно перемешивают, выливают в чашки, куда был внесен нистатин, и снова тщательно перемешивают с посевным материалом и нистатином (вращательным движением). В остальные чашки выливают дрожжевой агар, молочный агар и тщательно смешивают с посевным материалом и нистатином. После застывания агаровой среды чашки ставят в термостат крышкой вниз для выращивания при температуре 30 °С на 24-48 ч. Все операции по внесению в чашки отдельных ингредиентов производятся с соблюдением правил асептики в непосредственной близости от пламени горелки.
Через 24 ч после выдерживания в термостате подсчитывают колонии лейконостока на дрожжевом агаре с сахарозой. При этом нужно не пропустить момента, когда колонии имеют вид отдельных капель, позже колонии сливаются вместе, и их невозможно учесть. Через 48-72 ч подсчитывают количество гнилостных бактерий, так как к этому времени ясно обозначаются зоны просветления среды вокруг этих колоний на молочном агаре.
В результате подсчета количество бактерий выражают числом клеток (сотни, тысячи, миллионы, миллиарды) в 1 г прессованных дрожжей. Посторонние дрожжи выражают в процентах от общего количества дрожжей.
|
В 1 г дрожжей ЧК допускается: |
|
|
молочнокислых бактерий |
До 20 млн. |
|
лейконостока |
0 |
|
кишечной палочки |
0 |
|
гнилостных бактерий, не более |
500 |
|
посторонних дрожжей |
0 |
|
В 1 г дрожжей ЕЧК допускается: |
|
|
молочнокислых бактерий |
До 20 млн. |
|
лейконостока |
0 |
|
кишечной палочки |
0 |
|
гнилостных бактерий |
До 1000 |
|
посторонних дрожжей |
0 |
Зарубежные методы выявления посторонней, вредной для производства микрофлоры
Выявление бактериальной зараженности. Определение общего количества бактерий. Пробу бродящей жидкости из промежуточных стадий разводят стерильным 0,5%-ным физиологическим раствором в соотношении 1:10, 1:100 и 1:1000 и соответственно производят посевы внутрь питательной среды по 1 мл из перечисленных разведений. (Для подавления роста дрожжей применяют питательную среду с добавлением актидиона - см. приложение II.) Число колоний в чашках должно быть не менее 30 и не более 300.
Пробу прессованных дрожжей разводят физиологическим раствором в соотношении 1:100, 1:1000, 1:10000, 1:100000, 1:1000000 и соответственно проводят посевы. Из этих разведений вносят по 1 мл внутрь среды и по 0,1 мл на поверхность среды с актидионом.
Выросшие колонии подсчитывают сначала невооруженным глазом, затем с лупой или при помощи микроскопа со слабым увеличением (60-80 раз).
Выявление бактерий, вредных для производственного процесса и готовой продукции. Для выявления спорообразующих бактерий пробу, разведенную в соотношениях 1:10 и 1:100, выдерживают перед посевом 30 мин в водяной бане при 80 °C.
Выявление бактерий, протеолизирующих и вызывающих гидролиз крахмала. Производят посев из всех разведений на агаризованную среду с крахмалом и желатиновую среду (основой служит дрожжевая вода) по 0,1 мл на поверхность питательных сред. Посевной материал распределяют по поверхности среды при помощи шпателя Дригальского. Выращивание посевов на желатиновой среде ведут при комнатной температуре 48-72 ч, на среде с крахмалом - при температуре 30 °С в термостате 48-72 ч.
Колонии протеолизирующих бактерий разжижают желатину и слегка углубляются в среду; колонии гидролизующих крахмал бактерий образуют зону просветления.
Выявление клостридий сульфовосстановителей. Присутствие этих бактерий в дрожжах опасно, так как они разлагают сернистые соединения дрожжей с образованием сероводорода. Для того чтобы их обнаружить, производят посев на специальную среду (см. приложение II).
Пробу дрожжей разбавляют 1:10 физиологическим раствором и пастеризуют 10 мин при 80 °С, охлаждают и производят посев в питательную среду. Посевной материал вносят в три пробирки со средой во все убывающем количестве: в 1-ю пробирку 5 мл суспензии дрожжей; во 2-ю - 3 мл суспензии + 2 мл физиологического раствора; в 3-ю пробирку 2 мл суспензии + 3 мл физиологического раствора.
Среду и посевной материал тщательно перемешивают и добавляют в каждую по 1 мл стерилизованного парафинового масла. Все пробирки помещают в термостате при 37 °С на 48 ч.
В случае наличия сульфатредуцирующих бактерий в пробирках появляются линзообразные колонии, окруженные черной зоной.
Посев в элективную среду*. Одну каплю пробы из аппарата или одну петлю из середины кусочка прессованных дрожжей или из концентрата в воде вносят с соблюдением правил асептики в накопительную (элективную) ацетатную среду. Пробирку с посевом помещают в термостат при 30 °С и наблюдают в течение 5 сут за появлением мути или пленки на поверхности среды.
________________
* Метод используется для выявления очень незначительного количества клеток посторонних дрожжей, иногда не обнаруживаемых при высеве на чашке Петри.
Чем раньше появится пленка на поверхности среды, тем более инфицированным посторонними дрожжами является материал. Этот метод позволяет дать оценку степени зараженности посторонними дрожжами анализируемой пробы.
Если пленка появилась через 5 или большее число дней или совсем не появилась, это значит, что в исследуемом материале очень мало посторонних дрожжевых грибков или же их нет совсем. Если же пленка появилась через 3-4 дня, то дрожжи ЧК или ЕЧК можно считать удовлетворительными, пригодными для работы по удлиненным схемам. Появление пленки через 1-2 дня свидетельствует о том, что исследуемый материал содержит значительное количество посторонних дрожжей, дрожжи ЧК и ЕЧК неудовлетворительные и непригодные для работы по удлиненным схемам.
Контроль ферментативной активности дрожжей ЧК и ЕЧК
В оценку ферментативной активности входит определение подъемной силы, зимазной и мальтазной активности.
Определение подъемной силы методом всплывания шарика теста. Подъемная сила должна составлять 35-45 мин при определении ее по методике, описанной в действующем ГОСТе на дрожжи хлебопекарные.
На технических весах отвешивают навеску испытуемых дрожжей в количестве 0,31 г. К навеске добавляют 4,8 мл 2,5%-ного раствора поваренной соли, нагретого до 35 °С, и тщательно размешивают пестиком в фарфоровой ступке или чашке. К полученной дрожжевой эмульсии добавляют от 6,5 до 7,5 г (в зависимости от влажности) муки 85%-ного помола и быстро замешивают тесто, придавая ему форму шарика, который не должен прилипать к рукам.
Шарик опускают в стакан с водой температурой 35 °С и ставят в термостат при температуре 35 °С, заметив время, когда был опущен шарик в воду, и наблюдают за шариком до тех пор, пока он не всплывет к поверхности воды. Время всплывания шарика отмечают. Промежуток времени между опусканием шарика в воду и его всплыванием должен быть не более 8-10 мин у хороших дрожжей. Это время следует умножить на коэффициент 3,5, полученный эмпирически, для перевода на количество минут, найденное стандартным методом определения подъемной силы.
Пример. Время всплывания шарика 10 мин. 10·3,5=35 мин при определении стандартным методом.
Определение зимазной и мальтазной активности. Показатели зимазной и мальтазной активности, являющиеся суммарными показателями работы ферментов дрожжей и муки, в отличие от подъемной силы означают активность только дрожжевых ферментов.
Зимазная и мальтазная активность выражается временем, необходимым для выделения 10 мл диоксида углерода при сбраживании 20 мл 5%-ного раствора сахара прессованными дрожжами, взятыми в количестве 2,5% к объему среды (подробно метод описан на с.16).
Зимазная активность качественных маточных и засевных дрожжей должна составлять 30-40 мин, мальтазная активность - не превышать 80-90 мин.
Оценка стойкости ферментных систем дрожжей при хранении
Маточные дрожжи используют на заводах обычно не сразу после выработки, а хранят в холодильных камерах в течение нескольких недель. Поэтому дрожжи должны обладать способностью сохранять свою ферментативную активность длительное время.
Для выявления устойчивости ферментных систем предложено три показателя: стойкость при хранении, осмочувствительность, генеративная активность.
Оценка стойкости по изменению подъемной силы. Подъемная сила хранящихся дрожжей определяется периодически (через 5-10 сут) по стандартному методу (см. с.119) или ускоренным методом по "шарику" (см. с.27).
Если ферментные системы дрожжей устойчивы, то подъемная сила в процессе хранения не изменяется или ухудшается очень незначительно - на 1-2 мин за 10 сут. Если дрожжи нестойкие, то подъемная сила заметно ухудшается - на 5-10 мин за 10 сут хранения.
Определение и оценка осмочувствительности. Это свойство дрожжей является также хорошим показателем устойчивости ферментов дрожжей. Оно проявляется в средах с повышенным осмотическим давлением и выражается в понижении бродильной активности дрожжей. Это понижение может быть различным: сравнительно небольшим и очень значительным. Так, осмочувствительность дрожжей, полученных по воздушно-приточному способу, может колебаться от 0 до 75 мин (по данным Уайта).
Уайт предложил методику определения осмочувствительности, по которой этот показатель выражается разницей во времени между подъемной силой дрожжей в тесте без соли и в тесте с повышенной концентрацией ее - до 3,35%. Чем больше разница во времени, тем более высокой осмочувствительностью обладают дрожжи. Хорошо хранящиеся дрожжи должны обладать слабой осмочувствительностью (от 1 до 10 мин). Подробно метод описан на с.35.
Определение генеративной активности. Маточные дрожжи должны обладать высокой генеративной активностью - способностью дрожжевых клеток давать большое количество дочерних клеток за определенный промежуток времени (метод описан на с. 29).
КОНТРОЛЬ ПРОЦЕССА ВЫРАЩИВАНИЯ ДРОЖЖЕЙ В ТОВАРНОЙ СТАДИИ
Во всех стадиях выращивания товарных дрожжей: промежуточной, стадии выращивания засевных дрожжей, стадии выращивания товарных дрожжей, - периодически отбираются пробы из аппаратов для исследования под микроскопом. При этом определяют количество почкующихся клеток (в %) и количество посторонней микрофлоры (посторонних дрожжевых грибов и бактерий), наличие неправильно почкующихся клеток, наличие отмерших клеток.
Определение количества почкующихся клеток. Одну каплю взвеси дрожжей из аппарата смешивают на предметном стекле с одной каплей стерильной воды и накрывают покровным стеклом. Наблюдения проводят при увеличении в 600-800 раз.
Просматривают от пяти до десяти полей зрения, в каждом просчитывают общее количество клеток и отдельно количество клеток с почками. Выводят среднее и рассчитывают процентное содержание почкующихся клеток. При оптимальных условиях и соблюдении технологического режима количество почкующихся в различных стадиях технологического процесса клеток может быть различным в различные часы выращивания.
В промежуточной стадии количество почкующихся клеток составляет 30-40%, в засевной стадии (в начале процесса) - 80-90%, а к концу снижается до 30-40%.
В товарной стадии на первом часу 30-40% почкующихся клеток, к пятому часу количество их увеличивается до 80-90% и в конце процесса составляет 10-12%.
Определение количества посторонних дрожжевых грибов. Число клеток посторонних дрожжевых грибов считают в том же препарате, который был приготовлен для подсчета почкующихся клеток.
Клетки посторонних дрожжевых грибов отличаются от клеток основной культуры (сахаромицетов) по своей форме. Они обычно мельче, продолговатой формы, часто с несколькими вакуолями внутри клетки, имеются также сильно преломляющие свет одно-два тельца. Иногда клетки образуют микроколонии из нескольких почкующихся клеток. Подсчитывают отдельно клетки основной культуры и клетки посторонних дрожжей. Количество посторонних дрожжей (в процентах) рассчитывают по отношению к общему количеству клеток в поле зрения. Просчитывают как минимум пять полей зрения и выводят среднее.
Пример. В пяти полях зрения обнаружено 20 клеток посторонних дрожжей, всего в пяти полях зрения сосчитано 100 клеток дрожжей, следовательно, количество посторонних дрожжей составляет 20%. В конце товарной стадии такое число посторонних дрожжей допускается.
Определение наличия неправильно почкующихся клеток. Клетки с двумя, тремя почками, иногда уже отмершими, появляются при наличии в питательной среде дрожжерастильного аппарата веществ, угнетающих рост и размножение дрожжей (сахаромицетов). В нормальных условиях таких клеток не должно быть (рис.3 и 4).
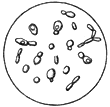
Рис.3. Проба из дрожжерастильного аппарата - правильно протекающий процесс: почкование правильное, примесь посторонних дрожжей незначительна
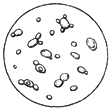
Рис.4. Проба из дрожжерастильного аппарата - неправильно протекающий процесс: клетки угнетены, почкование неправильное, значительная примесь посторонних дрожжей, есть отмершие клетки, окрашенные метиленовой синью
Определение количества отмерших клеток. Отмершие клетки появляются в среде дрожжерастильного аппарата при наличии в ней ядовитых веществ или при нарушениях температурного режима (повышение температуры) и при сдвигах рН в кислую сторону от оптимального диапазона (4,5-5,5).
Одну каплю среды из дрожжерастильного аппарата смешивают на предметном стекле с одной каплей разбавленной синьки (по Финку) (см. приложение II - краски) и через 1-2 мин просматривают препарат под микроскопом при увеличении в 600-800 раз. Отмершие клетки дрожжей окрасятся в синий цвет, так как клеточная оболочка и мембрана мертвых клеток не препятствуют проникновению краски. В живые клетки краска не проникает. При нормально протекающем процессе отмерших клеток не должно быть.
КОНТРОЛЬ ПРОЦЕССА ВЫДЕЛЕНИЯ ДРОЖЖЕЙ
Дрожжи выделяют из бражки на непрерывно действующих центрифугах-сепараторах. При помощи сепараторов дрожжи промывают водой, сгущают и в виде дрожжевого молока (густой взвеси) собирают в сборники (дрожжевые тарелки), откуда их подают на прессы или вакуум-фильтры или хранят в специальных сборниках при температуре 2-4 °С.
Контроль сводится к учету потерь дрожжей в этой стадии производства, к надзору за правильностью промывки и сохранностью дрожжевых концентратов.
Наиболее быстрым и удобным способом для учета потерь дрожжей является микроскопирование неокрашенных препаратов проб бражки и промывной воды, отходящих из сепараторов.
При нормальной работе сепараторов количество дрожжей в отходящей жидкости очень невелико: 1-2 клетки не в каждом поле зрения. Просматривают 5 полей зрения. В жидкости, отходящей от прессов, также не должно содержаться дрожжевых клеток, что устанавливают микроскопированием.
Величину потерь дрожжей в цехе выделения можно определить путем подсчета дрожжевых клеток в отходящей с сепараторов жидкости с помощью счетной камеры. Можно пользоваться камерой любой системы, применяемой для подсчета элементов крови при анализе крови (рис.5).
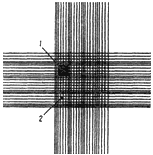
Рис.5. Счетная камера:
1 - большой квадрат; 2 - малый квадрат
Анализу подвергают жидкость, отходящую с сепараторов и прессов. Одну каплю этой жидкости помещают на середину камеры, где нанесена клетка, покрывают покровным стеклом (шлифованным) и притирают его до образования по краям стекла радужных Ньютоновых колец. Помещают на предметный столик микроскопа и считают клетки дрожжей при увеличении в 600-800 раз. Сначала определяют количество дрожжевых клеток в 1 мл исследуемой жидкости. Для пересчета необходимо знать, что каждый квадратный миллиметр поверхности дна камеры разделен на 25 больших квадратов и 400 малых, высота камеры, т.е. расстояние от дна камеры до нижней поверхности покровного стекла, составляет 0,1 мм. Отсюда объем одного маленького квадратика будет равен 1/4000 см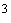 , или 1/4000000 мм. Считают клетки дрожжей в 80 маленьких квадратиках или в 5 больших, что равнозначно.
, или 1/4000000 мм. Считают клетки дрожжей в 80 маленьких квадратиках или в 5 больших, что равнозначно.
Пример пересчета. В 80 малых квадратах или в 5 больших найдено 15 клеток: 15·4000000/80=7·500000 клеток в 1 мл жидкости, в пересчете на 1 л жидкости - 7,5 млрд., т.е. около 1 г дрожжей в пересчете на прессованные. Следует учесть, что в 1 г прессованных дрожжей содержится от 8 до 10 млрд. дрожжевых клеток в зависимости от их размеров.
Определяют объем жидкости и подсчитывают общее количество дрожжей в этом объеме в килограммах. На основании полученных данных находят величину потерь в процентах по отношению к общему выходу дрожжей из затора или за сутки. Для правильного учета потерь отбирают пробы при первой, второй и третьей сепарациях затора или из общего желоба для отходящей жидкости. Лучше всего учитывать потери дрожжей за каждые 8 ч путем анализа средней пробы, отбираемой автоматически.
При правильной работе сепараторов и прессов потери дрожжей в этой стадии не превышают 0,1% по отношению к количеству дрожжей, вырабатываемых заводом.
АНАЛИЗ ПРЕССОВАННЫХ ДРОЖЖЕЙ
Для оценки степени зараженности готовой продукции посторонними микроорганизмами (дрожжевыми грибами и бактериями) применяются методы, приведенные ниже.
Чашечный метод
Этот метод может применяться в двух вариантах: упрощенном и усложненном для более углубленных исследований посторонней микрофлоры.
Упрощенный метод. Посев производится на поверхность питательной среды агара-сусла с мелом.
Разведение материала состоит в следующем. Комочек дрожжей при помощи петли, предварительно прокаленной и охлажденной, переносят в 10 мл стерильной водопроводной воды и тщательно размешивают. Стерильной пипеткой вносят 1-2 капли взвеси на поверхность ранее заготовленной в чашке Петри среды сусла-агара с мелом (чашка N 1), размазывают стерильным шпателем Дригальского и затем этим же шпателем распределяют остатки материала на поверхности чашек N 2 и 3 с той же средой (истощающий посев). Способ разливки среды в чашки описан на с.5. Засеянные чашки помещают в термостат при 30 °С на 48 ч. По прошествии этого срока на поверхности среды вырастают:
-
характерные колонии дрожжей основной культуры - сахаромицетов - круглые, выпуклые, с гладкой поверхностью и ровным краем, светло-серого или слегка желтоватого цвета;
-
колонии посторонних дрожжевых грибов - обычно из рода Кандида - разные, но хорошо отличимые от колоний сахаромицетов - крупные, плоские, матовые, сероватые с ворсинчатым краем или плоские с гладкой или слегка складчатой поверхностью сероватого цвета и др.;
-
колонии кислотообразующих бактерий, мелкие опалесцирующие, с зоной просветления вокруг себя;
-
спорообразующие бактерии - колонии сухие, плоские, слегка складчатые, с фестончатым краем, крупные, сероватые или слизистые, иногда с пузырьками газа.
Выросшие колонии подсчитывают на тех чашках, где их не более 50 и не менее 20, выводят среднее из двух-трех чашек и определяют процентное соотношение.
Пример. Выросло колоний сахаромицетов (среднее) 35, колоний посторонних грибов 10, кислотообразующих бактерий 7, спорообразующих бактерий 1. Производят два расчета.
Расчет соотношения основной культуры и посторонних дрожжевых грибов: общее количество колоний 35+10=45, отсюда 10·100/45=22,5%.
Расчет соотношения общего количества дрожжей и бактерий: общее количество дрожжей 45, бактерий 7+1=8, отсюда 8·100/53=15,1%. Всего 45+8=53, из них 13,2% кислотообразующих (7·100/53) и 1,9% спорообразующих бактерий (15,1-13,2).
В прессованных товарных дрожжах хорошего качества допускается наличие посторонних дрожжевых грибов не более 5-10%, кислотообразующих бактерий - не более 15-20%.
Усложненный метод. Используется в случаях замеченных отклонений в технологическом режиме производства, снижения качества готовой продукции (плохой подъемной силы и недостаточной стойкости при хранении). В этих случаях проводят более углубленные исследования - посев на несколько различных сред для выявления абсолютного количества клеток вредных микроорганизмов и их определения по группам (метод разработан Ленинградским отделением ВНИИХПа).
При исследовании товарных прессованных дрожжей пользуются схемой (табл.3).
Таблица 3
|
Группа микроор- |
Питательная среда |
Разведение 1 г дрожжей |
||||||
|
100 |
1000 |
10 тыс. |
100 тыс. |
1 млн. |
10 млн. |
100 млн. |
||
|
Культурные и посторонние дрожжи |
Сусло-агар |
- |
- |
- |
- |
- |
2 чашки |
2 чашки |
|
Посторонние дрожжи |
Сусло-агар |
- |
- |
- |
- |
- |
2 чашки |
2 чашки |
|
Синтетическая среда с лизином |
- |
- |
- |
- |
- |
2 чашки |
2 чашки |
|
|
Бактерии и лейконосток |
Дрожжевой агар с сахарозой и нистатином |
- |
- |
- |
2 чашки |
2 чашки |
2 чашки |
- |
|
Молочнокислые бактерии (лактобактерии с лейконостоком) |
Сусло-агар с мелом и нистатином |
- |
- |
- |
2 чашки |
2 чашки |
2 чашки |
- |
|
Гнилостные бактерии |
Молочный агар с нистатином |
2 чашки |
2 чашки |
2 чашки |
- |
- |
- |
- |
Дальнейший разбор посевов производится так же, как и при исследовании дрожжей ЧК и ЕЧК (см. с.20).
В доброкачественных дрожжах допускаются следующие нормы содержания посторонних микроорганизмов (расчет дан на 1 г прессованных дрожжей):
|
Молочнокислые бактерии |
До 100 млн. |
|
Лейконосток |
До 1 млн. |
|
Кишечная палочка |
0 |
|
Гнилостные бактерии |
5000 |
|
Дикие дрожжи |
До 10% |
Микрометод определения подъемной силы дрожжей (несахаромицетов)
Дрожжевые грибы (несахаромицеты), размножающиеся в процессе производства хлебопекарных дрожжей (посторонние дрожжи), имеют пониженную ферментативную активность, и примесь их к основной культуре влияет на качество прессованных и сушеных дрожжей, главным образом на подъемную силу.
Излагаемый ниже метод имеет своей целью определить степень вредности того или иного вида посторонних дрожжей, их влияния на подъемную силу готовой продукции (хлебопекарных дрожжей).
Метод заключается в следующем. Культуру посторонних дрожжей выделяют из прессованных дрожжей путем выделения отдельных колоний, выросших в чашках Петри при анализе дрожжей чашечным методом, на скошенный питательный сусло-агар в пробирки. После того как исследуемая культура вырастет на скошенном агаре, в пробирку наливают 1-2 мл стерильной водопроводной воды и сильно встряхивают. Несколько капель этой эмульсии наносят на поверхность застывшего сусла-агара в чашке Петри. Эмульсию размазывают по поверхности шпателем Дригальского и выращивают в термостате при 30 °С 48-72 ч, после чего налет дрожжей соскабливают химическим шпателем. Полученную дрожжевую массу подсушивают на фильтровальной бумаге до влажности около 75% и определяют подъемную силу методом всплывания шарика теста (по Островскому). Метод подробно описан на с.27.
При использовании этого метода получаются следующие данные по подъемной силе посторонних дрожжевых грибов, наиболее часто встречающихся в прессованных дрожжах, и чистой культуры сахаромицетов для сравнения:
|
Гриб |
Подъемная сила, мин |
|
Кандида гиллермондии |
64-90 |
|
Кандида крузеи |
74-160 |
|
Кандида брумптии |
Не поднимает теста |
|
Сахаромицеты (раса N 7) |
18-20 |
Для пересчета на стандартную подъемную силу полученные величины умножают на коэффициент 3,5, как и при определении подъемной силы прессованных дрожжей.
Определение осмочувствительности
Осмочувствительность - это свойство дрожжей снижать бродильную активность в средах с повышенным осмотическим давлением. Осмочувствительные хлебопекарные дрожжи медленнее поднимают тесто с повышенным содержанием соли и сахара.
Метод основан на сравнительной оценке подъемной силы при нормальном и повышенном осмотическом давлении, т.е. в тесте без соли и с повышенным содержанием соли.
Разница во времени, выраженная в минутах, характеризует степень осмочувствительности прессованных дрожжей. Осмочувствительность прессованных товарных дрожжей в зависимости от их качества приведена ниже.
|
Осмочувствительность, мин |
Оценка дрожжей |
|
От 1 до 10 |
Хорошая |
|
От 10 до 20 |
Удовлетворительная |
|
Более 20 |
Неудовлетворительная |
Наиболее быстрым и легко выполнимым является метод с использованием способа всплывания шарика теста (по Островскому).
Ход определения заключается в следующем. На технохимических весах отвешивают две навески дрожжей по 0,31 г каждая. К первой навеске добавляют 4,8 мл водопроводной воды, нагретой до 35 °С, и тщательно размешивают при помощи химического шпателя или пестика в фарфоровой ступке или чашке.
К полученной дрожжевой эмульсии добавляют от 6,5 до 7,5 г (в зависимости от влажности) муки 85%-ного помола и быстро замешивают тесто, придают ему форму шарика, который не должен прилипать к рукам. Шарик опускают в стакан с водой при температуре 32 °С и помещают в термостат при 32-34 °С. Подъемная сила характеризуется временем, прошедшим с момента опускания шарика в воду до момента его всплывания.
Ко второй навеске добавляют 4,8 мл 3,35%-ного раствора поваренной соли, нагретого до 35 °С, и поступают так же, как и с первой навеской.
Шарик, замешанный на воде без соли, всплывает быстрее, чем шарик с солевым раствором повышенной концентрации.
Примечание. Время подъема шарика в минутах следует умножить на коэффициент 3,5, полученный эмпирически, для перевода на количество минут, найденное стандартным методом - по подъему теста.
АНАЛИЗ СУШЕНЫХ ДРОЖЖЕЙ
Сушеные дрожжи выгодно отличаются от прессованных способностью длительно сохраняться в том случае, если они высушены до остаточной влажности 6-8% и при этом в них сохранены исходные ферментативные свойства. В герметической упаковке они могут сохранять активность в течение нескольких лет в условиях хранения при комнатной температуре.
Сохранность сушеных дрожжей обусловлена качеством прессованных дрожжей, предназначенных для высушивания, режимом сушки, герметичностью упаковки.
Определение количества отмерших клеток
Отобранную для анализа пробу сушеных дрожжей предварительно обводняют во влажной камере (чашке Петри, на крышке которой помещен увлажненный кружок фильтровальной бумаги) и выдерживают 1-2 ч.
Около 0,1 г вносят в пробирку со стерильным солодовым суслом плотностью 8% СВ по сахарометру. Пробирку ставят в термостат при температуре 30 °С на 4 ч. За это время все жизнеспособные клетки, содержащиеся в пробе, успевают выйти из состояния анабиоза и начинают почковаться.
Из подмоложенных описанным выше способом дрожжей приготовляют препарат, который окрашивают разбавленной синькой, приготовленной на буферной смеси (см. приложение II). Для этого каплю взвеси подмоложенных дрожжей смешивают на предметном стекле с каплей синьки (по Финку) и покрывают покровным стеклом. Препарат просматривают под микроскопом при увеличении в 600-800 раз. Отмершие клетки окрашиваются синькой в синий цвет, так как оболочки и мембраны отмерших клеток свободно пропускают краску; ожившие клетки остаются неокрашенными.
Просчитывают пять полей зрения, в каждом поле зрения подсчитывают общее количество клеток и отдельно - окрашенные. Вычисляют число отмерших клеток по отношению к общему числу клеток; материнскую клетку с неотделившейся почкой считают на одну клетку.
Пример. Всего клеток в поле зрения 106, из них отмерших 18. Отсюда 18·100/106=16,9 (округленно 17%). Активные сушеные дрожжи должны содержать не более 50% отмерших клеток.
Определение подъемной силы (ускоренный метод с использованием способа всплывания шарика теста)
Предварительно отвешивают две навески пшеничной муки 85%-ного помола (0,65 и 8 г) и ставят в термостат при 35 °С. Через 30 мин берут навеску сушеных дрожжей 0,16 г (на технохимических весах), смачивают 1,5 мл водопроводной воды и ставят в термостат при 35 °C на 30 мин для подмолодки.
В фарфоровую чашку с подмоложенными дрожжами вносят 4 мл 2,5%-ного раствора поваренной соли, который получают так: 4 г соли растворяют в 130 мл водопроводной воды. Затем в фарфоровую чашку добавляют 8 г подогретой муки и замешивают шарик. Шарик опускают в стакан с водой при температуре 32 °С и ставят в термостат при 35 °С. Подъемная сила характеризуется временем, прошедшим с момента опускания шарика в воду до момента его всплывания.
Подъемная сила активных сушеных дрожжей по шарику 20-21 мин, что соответствует 70-74 мин при определении стандартным методом.
КОНТРОЛЬ САНИТАРНОГО СОСТОЯНИЯ АППАРАТУРЫ
Контроль за санитарным состоянием дрожжевого завода, проводимый систематически, является одним из условий, обеспечивающих выпуск высококачественной продукции.
Посторонние микроорганизмы, размножающиеся на остатках питательной среды и дрожжей, в плохо вымытой и непродезинфицированной аппаратуре, попадают в готовую продукцию, а также могут размножаться в основной культуре дрожжей. Они нарушают правильное питание размножающихся дрожжей - сахаромицетов, а иногда и угнетают их рост. Нарушается правильное течение технологического процесса, и ухудшается качество готовой продукции, в основном подъемная сила и стойкость. Может снизиться и выход продукции с 1 ед. сырья.
Для постоянного поддержания высокого уровня санитарного состояния производства необходим регулярный контроль, позволяющий выявить очаги и источники инфекции и своевременно их ликвидировать.
Дрожжерастильные аппараты, сборники для мелассы, дрожжей и другие емкости контролируют после мойки, дезинфекции и пропаривания с последующей промывкой холодной водой.
В стерильную пробирку или колбу отбирают несколько миллилитров последней промывной воды. Воду просматривают под микроскопом. В хорошо вымытой аппаратуре при просмотре препарата из промывной воды под микроскопом может быть обнаружено не более 1-2 бактерий не в каждом поле зрения, а дрожжей не должно быть совсем.
В сомнительных случаях и при наличии инфекции на заводе производят анализ промывной воды чашечным методом (посев внутрь питательной среды 1 мл промывной воды). В промывной воде не должно быть обнаружено больше микроорганизмов, чем в воде, идущей для промывки.
Промывную воду можно проверить и другим способом. В чистые, протертые спиртом пробирки центрифуги наливают по 10-15 мл последней промывной воды и центрифугируют при частоте вращения 1500-2000 об/мин в течение 10 мин. Затем воду сливают и каплю осадка переносят на предметное стекло, накрывают предметным стеклом и микроскопируют. В десяти полях зрения должно быть не более 5-6 микроорганизмов, посторонние дрожжевые грибы не должны встречаться, особенно в дрожжерастильных аппаратах, где размножаются чистая культура, маточные дрожжи ЧК и ЕЧК.
Качество дезинфекции аппаратуры проверяют в смыве с внутренней поверхности аппаратов, делают мазок или соскоб. На обследуемую поверхность накладывают рамку (шаблон) площадью 10, 25 или 100 см . Предварительно необходимо продезинфицировать рамку в пламени горелки и плотно приложить к обследуемой поверхности. Стерильным ватным тампоном, увлажненным стерильной водой, тщательно протирают поверхность, ограниченную рамкой, затем тампон бросают в ту же пробирку или колбу со стерильной водой, в которой он был увлажнен ранее. Жидкость взбалтывают вместе с тампоном и просматривают под микроскопом или производят посев в чашку Петри для определения общего количества микроорганизмов внутренней поверхности аппарата.
. Предварительно необходимо продезинфицировать рамку в пламени горелки и плотно приложить к обследуемой поверхности. Стерильным ватным тампоном, увлажненным стерильной водой, тщательно протирают поверхность, ограниченную рамкой, затем тампон бросают в ту же пробирку или колбу со стерильной водой, в которой он был увлажнен ранее. Жидкость взбалтывают вместе с тампоном и просматривают под микроскопом или производят посев в чашку Петри для определения общего количества микроорганизмов внутренней поверхности аппарата.
При тщательной мойке и дезинфекции количество микроорганизмов в жидкости не будет значительно превышать количество их в воде, идущей в производство. Под микроскопом в препарате дрожжей не должно быть совсем, а бактерий - 1-2 не в каждом поле зрения.
Качество мойки и дезинфекции трубопроводов и шлангов оценивают путем отбора нескольких проб последней промывной воды в стерильную посуду.
Воду исследуют так же, как это описано выше при контроле чистоты аппаратуры, и предъявляют к ней те же требования.
Сепараторы, фильтр-прессы, вакуум-фильтры, формовочные машины, воздуховоды и др. проверяют путем протирания ватным тампоном, смоченным стерильной водой, участков, соприкасающихся с продукцией или труднодоступных для мойки. Тампон держат пинцетом, предварительно прокаленным в пламени горелки. Затем этот тампон опускают в колбу со стерильной водой, сильно взбалтывают и микроскопируют. В препарате при просмотре нескольких полей зрения дрожжей не должно быть совсем, а бактерий 1-2 не в каждом поле зрения.
Чистоту стен и пола в производственных помещениях определяют путем соскоба или смыва с поверхности стены или пола при помощи шаблона. Соскоб переносят в пробирку или колбу со стерильной водой, тщательно взбалтывают и просматривают под микроскопом. В случае необходимости для выявления источников инфекции производят посев материала в чашки Петри. Требования те же, что и для аппаратуры.
КОНТРОЛЬ ВОДЫ
Санитарно-гигиеническую оценку питьевой и производственной воды осуществляют периодически согласно действующему ГОСТу в специальных лабораториях Государственной санитарной инспекции, но некоторые качественные показатели производственной воды определяют в заводской лаборатории. При этом учитывают общее количество микроорганизмов и периодически определяют степень пригодности воды для производственных целей.
Пробы воды отбирают из заводских водоисточников в стерильную посуду в количестве около 1 л, закрывают ватной пробкой с бумажным колпачком.
При взятии пробы из крана, трубы или колодца с насосом кран или выпускную трубу хорошо обжигают паяльной лампой или пламенем горящей ваты, смоченной спиртом, и через полностью открытый кран спускают воду в течение 10 мин. При отборе пробы горлышко посуды следует обжечь. Отобранные пробы воды перевозят охлажденными до 5 °С. Анализ воды производят тотчас или не позже чем через 2 ч после отбора пробы.
В воде определяют общее количество микроорганизмов в 1 мл. Санитарную оценку воды - определение коли-титра и коли-индекса - производят в санитарно-бактериологической лаборатории.
Для технологических целей и мойки аппаратуры должна использоваться водопроводная вода, прошедшая очистку, или вода из артезианских скважин, из глубоких водоносных слоев.
Кормы по содержанию микроорганизмов и по санитарным показателям следующие: общее количество микроорганизмов в 1 мл не более 100; коли-титр не ниже 300 (наименьший объем воды, в котором обнаружена кишечная палочка, равен 300 мл); коли-индекс 1-3 (в 1 л воды должно содержаться не более 1-3 клеток кишечной палочки).
Не разрешается использовать для производства воду из открытых водоемов (рек, прудов, озер) без очистки.
Определение общего количества микроорганизмов чашечным методом
Из пробы воды отбирают стерильной градуированной пипеткой 1 мл и вносят на дно стерильной чашки Петри, туда же выливают 10-15 мл расплавленного мясопептонного агара температурой 44-46 °С. Питательную среду тщательно перемешивают с посевным материалом при помощи легких вращательных движений, следя за тем, чтобы среда не попадала на борта чашки и в толще агара не образовывалось пузырьков воздуха.
Посев выращивают в термостате при температуре 37 °С, выросшие колонии подсчитывают через 24 ч. В случае необходимости применяют разведение материала стерильной водопроводной водой с таким расчетом, чтобы на чашке выросло не более 300 и не менее 30 колоний, для этого производят посев из разных разведений.
Если на чашке Петри выросло более 300 колоний и не было взято других разведений и анализ повторить нельзя, то допускается подсчет колоний в части чашки, но не менее чем на 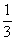 всей площади чашки.
всей площади чашки.
Определение степени пригодности воды для дрожжевого производства (по Вихману)
В воде, используемой для дрожжевого производства, имеет значение не только количество микроорганизмов, нормируемое согласно ГОСТу, но и видовой состав их. Особенно большое значение имеют те микроорганизмы, которые могут развиваться в кислых сахарных средах в процессе производства дрожжей.
Поэтому воду, используемую для производственных целей, оценивают по специально разработанным коэффициентам Вихмана. Вода с коэффициентом 0-20 считается очень хорошей, 20-40 - хорошей, 40-60 - удовлетворительной, 60-80 - плохой, 80-100 - непригодной.
Коэффициенты Вихмана характеризуют степень размножения бактерий воды в стерилизованном сусле в течение 5 дней при температуре 30 °С. Размножение бактерий обнаруживается тем быстрее, чем больше их в исследуемой воде.
Для проведения анализа в четыре колбочки наливают по 50 мл неохмеленного солодового сусла и стерилизуют в течение 20 мин при избыточном давлении 50 кПа. Перед посевом воды колбочки нумеруют и охлаждают. В охлажденную среду при помощи стерильной градуированной пипетки наливают исследуемую воду в следующих количествах:
|
Номер колбочки |
1 |
2 |
3 |
4 |
|
|
Количество воды, мл |
1,0 |
0,75 |
0,5 |
0,25 |
Засеянные колбочки помещают в термостат при температуре 30 °С и наблюдают ежедневно в течение 5 сут, отмечая номер колбочки и день, когда в ней появится муть, т.е. видимый рост микроорганизмов. Для оценки воды применяют условные коэффициенты для каждого дня.
|
День наблюдения и появления мути |
1-й |
2-й |
3-й |
4-й |
5-й |
|
Коэффициент мути |
10 |
8 |
6 |
4 |
2 |
На эти коэффициенты умножают номер колбочки, в которой наблюдалось помутнение.
В табл.4 приведен пример определения качества воды в процентах.
Таблица 4
|
Номер колбы |
Количество |
День, на которым наблюдалось помутнение |
Коэф- |
Результат определения качества, воды, % |
|
1 |
1 |
1-й |
10 |
1·10=10 |
|
2 |
0,75 |
3-й |
6 |
2·6=12 |
|
3 |
0,5 |
4-й |
4 |
3·4=12 |
|
4 |
0,25 |
На 5-й день помутнения |
0 |
4·0=0 |
Суммируя результат, получаем коэффициент изменения 34, на основании чего производится оценка воды (см. выше) - хорошая.
КОНТРОЛЬ ВОЗДУХА
Воздух производственных помещений и особенно вдуваемый в дрожжерастильные аппараты может быть источником инфекции, так как содержит самые разнообразные микроорганизмы, попавшие в него из почвы, с растений и др. Количество микроорганизмов в воздухе городов может достигать десятков и сотен тысяч в 1 м в зависимости от времени года, степени запыленности и др.
В процессе аэрации в дрожжевом производстве ежечасно расходуют тысячи кубометров воздуха, несущего десятки миллионов микроорганизмов.
Для борьбы с этой инфекцией воздух пропускают через воздуховоды, снабженные специальными фильтрами, улавливающими микроорганизмы, что значительно сокращает содержание их в воздухе (в 3-4 раза). Однако все же в дрожжерастильные аппараты попадает значительное количество посторонних микроорганизмов, часть которых размножается в процессе производства.
Для определения количества микроорганизмов в воздухе различных производственных помещений можно пользоваться седиментационным методом, достаточно точным и не требующим специальной аппаратуры.
Ход анализа заключается в следующем. Заготавливают две стерильные чашки Петри с питательной агаровой средой (сусло с мелом или дрожжевая вода с 4% сахарозы). Затем чашки завертывают в ту же стерильную бумагу, в которую они были завернуты при стерилизации, вносят в помещение, где исследуют воздух, и ставят на пол на развернутую бумагу. Крышки сдвигают так, чтобы вся поверхность агаровой пластинки была открыта полностью. Чашки оставляют открытыми в течение определенного периода времени (период экспозиции). Этот период должен быть кратным 5, т.е. 5, 10 или 15 мин, после чего крышку закрывают и чашки помещают в термостат при 30 °С крышкой вниз. Подсчет выросших колоний производят через 36-48 ч.
Для определения количества микроорганизмов в 1 м воздуха пользуются правилом и формулой, предложенными Омелянским. Автор метода подметил, что на площадь чашки Петри (100 см) оседает в течение 5 мин столько микроорганизмов, сколько их содержится в 10 л воздуха. Предполагается, что из каждой клетки, упавшей на поверхность среды, образуется колония. Расчет ведут по формуле 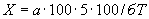 , где
, где  - количество микроорганизмов в 1 м воздуха; - число колоний, выросших на чашке Петри (среднее из трех); - площадь чашки Петри, см;
- количество микроорганизмов в 1 м воздуха; - число колоний, выросших на чашке Петри (среднее из трех); - площадь чашки Петри, см;  - время экспозиции; 100 - пересчет площади, взятой чашки Петри на 100 см; 100 - пересчет на 1 м; 5 - экспозиция чашки по Омелянскому, мин.
- время экспозиции; 100 - пересчет площади, взятой чашки Петри на 100 см; 100 - пересчет на 1 м; 5 - экспозиция чашки по Омелянскому, мин.
Пример. На чашке Петри площадью 76 см после выдержки в течение 5 мин выросло через 48 ч 12 колоний, следовательно, в 1 м воздуха будет содержаться 12·100·5·100/76·5=1580 микроорганизмов (округленно).
Воздух считается чистым, если в нем содержится не более 500 микроорганизмов в 1 м, кроме того, в воздухе производственных помещений не должно быть посторонних дрожжей.
Воздух, подаваемый в дрожжерастильные аппараты, анализируется следующим методом: определенный объем пропускается через стерильную воду, а затем производится посев этой воды в чашки Петри.
Для выполнения этого анализа необходим прибор, состоящий из реометра склянки Дрекселя или простой широкогорлой склянки 1 общей вместимостью 0,5-0,9 л, закрытой резиновой пробкой с двумя отверстиями, через которые пропущены две стеклянные трубки - подводящая 2 и отводящая 3 (рис.6). (Подводящая трубка доходит до дна склянки и оканчивается стеклянным шариком с дырочками, другая - отводящая, короткая, заканчивается непосредственно под пробкой; обе в верхней части загнуты под прямым углом.)
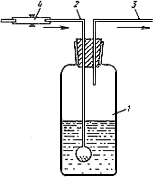
Рис.6. Прибор для микробиологического анализа воздуха
Перед употреблением прибор стерилизуют в автоклаве. Трубки с пробкой завертывают в бумагу и стерилизуют при 50 кПа в течение 30 мин. В склянку наливают 300 мл водопроводной воды, закрывают ватной пробкой и стерилизуют при 100 кПа в течение 1 ч.
Перед анализом прибор собирают, как указано на рис.6. Подводящую трубку соединяют с воздуховодом (необходимо специальное устройство для забора воздуха), к отводящей присоединяют реометр и при помощи зажима 4
регулируют количество воздуха, проходящего через стерильную воду. Предполагается, что микроорганизмы, содержащиеся в воздухе, будут задержаны водой. В течение 10-5 мин через прибор пропускают 100-120 л воздуха. Далее производят посев воды внутрь агаризованной питательной среды (дрожжевая вода с 4% сахарозы или солодовое сусло плотностью 8% СВ). Вносят по 1 мл воды в 4-5 чашек Петри, смешивают со средой и выращивают при 30 °C в течение 36-48 ч. Выросшие колонии считают на всех чашках Петри и полученные результаты пересчитывают на 1 м воздуха.
Пример. В 5 чашках выросло 8 колоний, это значит, что в 5 мл содержится 8 микроорганизмов (во всем объеме воды в склянке 300 мг). Это составит 8·300/5=480 микроорганизмов в 200 л воздуха, пропущенного через воду, следовательно, в 1 м содержится 480·5=2400 микроорганизмов.
В типовой дрожжерастильный аппарат подается ежечасно до 6000 м воздуха, следовательно, 2400·6000=24400000 посторонних микроорганизмов вносится в дрожжерастильный аппарат ежечасно при слабой очистке воздуха.
Если воздух, вдуваемый в аппараты, хорошо очищается, то в нем не должно содержаться посторонних дрожжевых грибов, спор и конидий плесеней.
АНАЛИЗ СОСТАВА СТОЧНЫХ ВОД
Этот метод уточнен применительно к дрожжевому производству Т.И.Тарутиной.
Сточные воды дрожжевых заводов состоят из отработанного мелассового сусла и промывных вод, предназначенных для охлаждения и промывания выделенных дрожжей. Они содержат значительное количество органических и неорганических веществ.
Эквивалентом степени загрязненности сточных вод органическими веществами является количество кислорода, израсходованного в определенный промежуток времени на аэробное биохимическое разложение органических веществ, содержащихся в сточной воде, - биологическое потребление кислорода (БПК).
Для определения БПК необходимо предварительно определить ХПК, т.е. химическую потребность в кислороде. Ниже приведен метод определения этого показателя.
Определение химической потребности в кислороде
Количество кислорода, требуемое для окисления органических веществ сточной воды до диоксида углерода, воды и аммиака, называют химической потребностью в кислороде. Наиболее полное окисление достигается обработкой проб бихроматом калия в присутствии большого количества серной кислоты.
При кипячении в 50%-ной по объему серной кислоте бихромат калия окисляет большинство органических веществ и некоторые неорганические вещества. Для большей полноты окисления органических веществ прибавляют в качестве катализатора сульфат серебра. Мешающее влияние хлоридов исключают, вводя в раствор сульфат ртути (II). После окисления избыток бихромата калия определяют титрованием раствором соли Мора.
Ход определения заключается в следующем. В круглодонную колбу отбирают 1-5 мл сточной воды, прибавляют 25 мл 0,25 н. раствора бихромата калия KСrО и 0,4 г сульфата ртути. Одновременно следует ввести 5 мл концентрированной серной кислоты для лучшего растворения сульфата ртути (II), 0,4 г сульфата серебра, стеклянные шарики или кусочек пемзы. Смесь перемешивают и осторожно приливают к ней 30 мл концентрированной серной кислоты (сумма объема пробы и объема бихромата калия), после чего вставляют в колбу пришлифованный обратный холодильник и кипятят 2 ч. Затем смесь охлаждают, отсоединяют холодильник, смывают внутреннюю и нижнюю части холодильника дистиллированной водой (50 мл) и снова охлаждают смесь. Далее содержимое холодильника переносят в коническую колбу на 500 мл, смывают дистиллированной водой (50 мл), прибавляют 2-3 капли раствора ферроина или 5 капель раствора
и 0,4 г сульфата ртути. Одновременно следует ввести 5 мл концентрированной серной кислоты для лучшего растворения сульфата ртути (II), 0,4 г сульфата серебра, стеклянные шарики или кусочек пемзы. Смесь перемешивают и осторожно приливают к ней 30 мл концентрированной серной кислоты (сумма объема пробы и объема бихромата калия), после чего вставляют в колбу пришлифованный обратный холодильник и кипятят 2 ч. Затем смесь охлаждают, отсоединяют холодильник, смывают внутреннюю и нижнюю части холодильника дистиллированной водой (50 мл) и снова охлаждают смесь. Далее содержимое холодильника переносят в коническую колбу на 500 мл, смывают дистиллированной водой (50 мл), прибавляют 2-3 капли раствора ферроина или 5 капель раствора 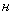 -фенилантраниловой кислоты и титруют избыток бихромата калия титрованным раствором соли Мора до изменения окраски индикатора.
-фенилантраниловой кислоты и титруют избыток бихромата калия титрованным раствором соли Мора до изменения окраски индикатора.
Параллельно проводят холостой опыт с 1-5 мл дистиллированной воды (вместо соответствующего объема сточной воды).
Бихроматную окисляемость (в мг О/л) вычисляют по формуле
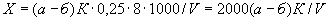 ,
,
где , - объем раствора соли Мора, израсходованный соответственно на холостой опыт и титрование пробы, мл; 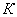 - поправочный коэффициент для приведения концентрации раствора соли Мора к точно 0,25 н.;
- поправочный коэффициент для приведения концентрации раствора соли Мора к точно 0,25 н.; 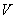 - объем пробы, взятый для определения, мл; 8 - эквивалент кислорода.
- объем пробы, взятый для определения, мл; 8 - эквивалент кислорода.
Обычно ХПК сточных вод производства дрожжей колеблется в пределах 5000-10000 мг О/л.
Определение биохимического потребления кислорода методом разведения
Сточную воду смешивают с разбавляющей водой, смесь до предела насыщают растворенным кислородом (встряхиванием), разливают в инкубационные склянки (рис.7) и тщательно герметизируют.

Рис.7. Калибровочная склянка для определения БПК в сточных водах
Склянки оставляют в термостате при температуре 20 °С на 5 сут или более. За счет органических веществ сточной воды развивается сообщество микроорганизмов, на что расходуется растворенный кислород.
Величина уменьшения кислорода в склянке, умноженная на степень разведения, дает численную величину БПК.
Необходимое разбавление подсчитывают ориентировочно по результатам определения бихроматной окисляемости (ХПК). Условно принимают, что биологическое потребление кислорода составляет 50% химического потребления кислорода, а так как в воде после инкубации при правильно взятом разбавлении должно остаться 8-10 мг/л кислорода, то величину ХПК делят на 4 или 5. Полученный результат показывает, во сколько раз надо разбавить анализируемую воду.
Пример. ХПК сточной воды составляет 6000 мг О/л. Для того чтобы получить в исследуемой воде 8-10 мг/л кислорода, необходимо следующее ее разбавление: 6000/5=1200. Округленно 1000.
Сточные воды дрожжевых заводов, как и все промышленные воды, как правило, содержат незначительное количество микроорганизмов, поэтому требуется искусственное заражение разбавляющей воды бактериальной культурой, адаптированной к утилизации исследуемых веществ.
Приготовление культуры микроорганизмов для заражения. Анализируемую сточную воду разбавляют водопроводной водой в 10-100 раз, прибавляют биогенные реактивы (см. ниже) и оставляют в сосуде, закрытом ватной пробкой, на 2-3 дня. Появление мути или пленки указывает на развитие микрофлоры (проверяют под микроскопом). Микрофлору переносят в пробу анализируемой сточной воды, разбавляют в 5-20 раз и после дальнейшего развития флоры отбирают 1 мл этой воды на 1 л разбавляющей.
Приготовление разбавляющей воды. Дистиллированную воду для приготовления разбавляющей воды насыщают кислородом путем отстаивания ее в течение суток в открытом виде при комнатной температуре. На 1 л такой дистиллированной воды добавляют растворы реактивов в последовательности, указанной ниже.
1. Сульфат аммония (NH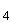 )SO (500 мг/л) - 5 мл.
)SO (500 мг/л) - 5 мл.
2. Сернокислый магний MgSО (50 мг/л) - 0,2 мл.
3. Сода (двууглекислый натр NaHCO) - 0,3 г.
4. Культура микроорганизмов для заражения (см. выше) - 1 мл.
5. Фосфатный буферный раствор, состоящий из Са(НРО) - 25 мг и KНРО - 250 мг в 1 л дистиллированной воды. Такой раствор добавляется в количестве 0,4 мл.
Разбавляющую воду приготовляют в день ее использования.
Ход определения заключается в следующем. В мерную колбу вместимостью 1 л наливают сифоном (чтобы в колбу не попали пузырьки воздуха) разбавляющую воду до половины колбы, прибавляют 1 мл исследуемой воды и снова доводят до метки разбавляющей водой. Закрыв колбу пробкой, тщательно перемешивают ее содержимое.
Разбавленную описанным способом сточную воду разливают для инкубации в пять калиброванных склянок вместимостью 150-200 мл, наполняя их доверху, и закрывают притертыми пробками так, чтобы не оставалось пузырьков воздуха. Затем эту же воду наливают в колпачки от склянок и, перевернув склянки вверх дном, вставляют их в колпачки, вытесняя из них воду так, чтобы не осталось пузырьков воздуха. Так же заполняют другие пять склянок разбавляющей водой. Каждой пробе анализируемой жидкости соответствует одна склянка с разбавляющей водой.
В одной из пяти склянок с анализируемой пробой и в одной из пяти склянок с разбавляющей водой сразу же определяют растворенный кислород. Все остальные склянки (8 шт.) помещают в термостат при 20 °С (±1°). Через 5, 10, 15, 20 сут от начала инкубации вынимают из термостата по одной склянке с анализируемой водой и по одной склянке с разбавляющей водой и определяют в них растворенный кислород и содержание нитритов.
Нитриты определяют (по Гриссу) в воде, налитой в колпачок склянки, который снимают так же, как надевали, т.е. перевернув склянку вверх дном. Если в пробе начался процесс нитрификации, что обнаруживается по появлению в воде нитритов концентрацией, превышающей 0,1 мг/л, то дальнейшее определение БПК не проводят. Если на пятые сутки появляются следы нитритов, то следующее определение проводят через 7-8 сут.
Химическое определение растворенного кислорода выполняют по методу Винклера. Этот метод основан на способности растворенного кислорода переводить марганец с валентностью +2 в марганец с валентностью +4, последний, реагируя с йодидом калия, выделяет из него свободный йод, который оттитровывается гипосульфитом натрия. Результат определения пересчитывают в эквиваленты кислорода и выражают в миллиграммах О на литр.
По истечении определенного инкубационного периода в анализируемой пробе сточной воды определяют растворенный кислород. В склянку с анализируемой водой прибавляют 1 мл раствора сульфата марганца (II) или хлорида марганца (II), погружая наполненную пипетку до дна склянки. Другой пипеткой прибавляют 1 мл раствора Винклера (гидроксид калия или натрия с йодидом калия), погружая кончик пипетки под уровень пробы в горлышко кислородной склянки.
Склянку осторожно закрывают так, чтобы под пробкой не образовывались пузырьки воздуха, и содержимое хорошо перемешивают, переворачивая склянку, до образования хлопьевидного осадка.
Прозрачный раствор над осадком сифонируют. К осадку в склянке по стенке приливают 5 мл разбавленной (1:4) серной кислоты и перемешивают. Содержимое склянки сливают в коническую колбу. Пробку и склянку ополаскивают несколько раз водой и все сливают в ту же коническую колбу, куда приливают 2 мл раствора йодида калия (15%), и содержимое вновь перемешивают. Через 5 мин выделившийся йод титруют 0,01 н. раствором гипосульфита натрия до светло-желтой окраски. Прибавляют раствор крахмала и продолжают титрование до обесцвечивания.
БПК разбавляющей воды вычисляют по формуле
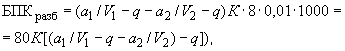
где 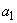 ,
,  - объем 0,01 н. раствора гипосульфита натрия, израсходованный на титрование жидкости при определении в ней содержания кислорода соответственно до инкубации и после инкубации, мл;
- объем 0,01 н. раствора гипосульфита натрия, израсходованный на титрование жидкости при определении в ней содержания кислорода соответственно до инкубации и после инкубации, мл; 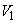 ,
, 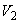 - вместимость склянок, в которых проводилось определение кислорода соответственно до инкубации и после инкубации, мл;
- вместимость склянок, в которых проводилось определение кислорода соответственно до инкубации и после инкубации, мл; 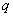 - объем реактивов (кроме кислоты), прибавленный в склянку при определении кислорода, мл; - поправочный коэффициент для приведения концентрации раствора гипосульфита натрия к точно 0,01 н.; 8 - эквивалент кислорода; 0,01 - нормальность раствора гипосульфита натрия.
- объем реактивов (кроме кислоты), прибавленный в склянку при определении кислорода, мл; - поправочный коэффициент для приведения концентрации раствора гипосульфита натрия к точно 0,01 н.; 8 - эквивалент кислорода; 0,01 - нормальность раствора гипосульфита натрия.
БПК анализируемой воды вычисляют по формуле
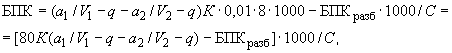
где  - объем сточной воды, взятой для анализа и разбавленный затем в мерной колбе до 1 л, мл.
- объем сточной воды, взятой для анализа и разбавленный затем в мерной колбе до 1 л, мл.
БПК стоков дрожжевого завода подвергается сильным колебаниям в течение суток. В среднем величина этого показателя колеблется от 1000 до 5000 мг О, тогда как на городские очистные сооружения принимаются стоки с БПК не выше 500 мг О/л.
ОПРЕДЕЛЕНИЕ УСТОЙЧИВОСТИ ДРОЖЖЕЙ К МЕЛАССЕ
Принцип этого метода заключается в том, чтобы определить степень чувствительности дрожжей производственной расы к вредным веществам мелассы или к недостатку каких-либо веществ. Сущность метода состоит в том, что дрожжи исследуемой культуры засеваются в стерильную мелассу плотностью 5% СВ.
Наблюдение за размножением дрожжей ведется в условиях ежесуточных пассажей на свежую среду, приготовленную из той же мелассы. Критерием является количество пересевов, которое способны выдержать дрожжи на данной мелассе. На мелассе нормального состава одни расы дрожжей выдерживают 6 пересевов, другие - 3, третьи - 1-2.
Чем выше чувствительность дрожжей различных рас к наличию в мелассе вредных веществ или к недостатку каких-либо ростовых веществ, тем быстрее падает способность дрожжей к размножению в процессе пересевов. Как показали наблюдения, шести пересевов достаточно для того, чтобы считать дрожжи устойчивыми к мелассе.
Подготовка мелассной среды. Мелассу в количестве 25-30 г разводят водой до плотности 5% СВ, разливают в пробирки по 10 мл в каждую и стерилизуют при 50 кПа 20 мин.
Подготовка дрожжей для засева и засев их в мелассу. Исследуемую культуру дрожжей с косого агара засевают в жидкое солодовое сусло плотностью 8% СВ и выдерживают в термостате при 30 °С 24 ч. Затем стерильно сливают верхний слой сусла и одну каплю осадка пипеткой вносят в пробирку со стерильной мелассой.
Пробирку ставят в термостат при 30 °С. Через 24 ч содержимое пробирки взбалтывают, одну каплю переносят в свежую (вторую) пробирку со стерильной мелассой и снова ставят в термостат на 24 ч, затем в третью и т.д. до 5-6 пересевов. Одновременно ведут наблюдение под микроскопом за количеством дрожжей, вырастающих в мелассе: считают количество клеток в пяти полях зрения.
При наличии роста дрожжей в поле зрения микроскопа должно быть не менее 4-5 клеток. При очень хорошем росте дрожжей в поле зрения микроскопа может наблюдаться до 20-30 клеток.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ БИОТИНА
Биотин - витамин Н, входящий в группу витаминов В, является стимулятором роста дрожжей. Чтобы обеспечить нормальную скорость роста и почкование дрожжей, биотин должен находиться в питательной среде в достаточном количестве - не менее 200 мг на 1 т сырья.
В свекловичной мелассе биотина, обычно, недостаточно для быстрого накопления биомассы, поэтому в мелассу добавляют вещества, содержащие биотин. Важно знать, сколько биотина содержится в перерабатываемом сырье и других продуктах (растительных экстрактах, автолизате дрожжей и др.).
Содержание биотина в мелассе характеризует ее качество, степень пригодности для дрожжевого производства (табл.5).
Таблица 5
|
Качество мелассы |
Количество биотина |
|
|
мкг/г |
мг/т |
|
|
Хорошее |
0,25-0,18 |
250-180 |
|
Среднее |
0,15-0,08 |
150-80 |
|
Неудовлетворительное |
0,05-0,03 |
50-30 |
Для определения биотина в мелассе, а также в растительных экстрактах, автолизате и других активаторах роста применяют микробиологический метод с использованием в качестве тест-организма дрожжи-сахаромицеты, например Saccharomyces cerevisiae, раса N 7.
Сахаромицеты очень чувствительны к наличию биотина в среде; рост их соответственно интенсифицируется с увеличением количества биотина в среде, содержащей полный набор всех остальных витаминов, необходимых для нормальной жизнедеятельности дрожжей.
В колбочку с синтетической средой, содержащей определенное количество исследуемого материала, засевают определенное количество дрожжей и выращивают в оптимальных условиях. По интенсивности роста, т.е. по количеству выросших дрожжей, судят о количестве биотина, сравнивая с накоплением дрожжей в стандартных растворах.
Количество анализируемого вещества необходимо брать с таким расчетом, чтобы содержание биотина в навеске не превышало 0,003-0,005 мкг, так как при большом количестве результаты получаются неточными. Это наблюдение подтверждается также и формой стандартной кривой: после дозы 0,01 мкг она имеет очень пологую форму, количество выросших клеток почти не изменяется, несмотря на увеличение дозы биотина.
Таблица 6
|
Номер колбы |
Навеска исследуемого вещества, г |
Количество дрожжей, млн./мл |
Количество биотина, мкг |
|
|
по стандартной шкале |
в 1 г исследуемого материала |
|||
|
1 |
0,15 |
45,0 |
0,01 |
0,066 |
|
2 |
0,1 |
39,5 |
0,0075 |
0,075 |
|
3 |
0,05 |
23,0 |
0,0035 |
0,070 |
|
Среднее |
0,070 |
|||
Наиболее удобные навески мелассы 0,1 и 0,005 г. Для получения таких навесок следует отвесить на технических весах 5 г мелассы (с точностью до 0,01 г) в стерильной градуированной пробирке (10 мл), затем стерильной дистиллированной водой довести до метки и тщательно размешать. Из этого раствора при помощи стерильной градуированной пипетки вносят 0,3; 0,2; 0,1 мл в три пробирки с заготовленной, как описано (см. приложение III), синтетической средой полного состава, но без биотина и затем засевают дрожжами (1 млн. клеток), доводят до метки, переливают в колбочки, ополаскивают 1 мл стерильной воды, доводят тем самым до объема 11 мл и ставят в термостат при 30 °С на 20-24 ч.
Количество выросших клеток подсчитывают в счетной камере или определяют по мутности нефелометром и сравнивают полученные результаты со стандартной кривой. Количество биотина в данной навеске пересчитывают на 1 г.
Для получения точных результатов анализ производят в трех повторностях с разным количеством исследуемого вещества и вычисляют среднюю величину (табл.6).
Для проведения анализа требуется специально обработанная посуда и синтетическая среда (способ обработки посуды и приготовление синтетической среды см. в приложении II).
Тест-организм - культуру дрожжей Saccharomyces cerevisiae, раса N 7 - поддерживают на сусле-агаре (скошенном), пересевают не реже одного раза в месяц с предварительной подмолодкой в жидком солодовом сусле в течение 18-20 ч с последующим посевом на скошенный сусло-агар. Выращивание культуры и подмолодку ведут при 30 °С, хранят при комнатной температуре.
Анализ начинают с приготовления стандартного ряда. В штатив помещают девять градуированных пробирок вместимостью 10 мл и по две-три таких же пробирки на каждый образец исследуемого вещества. В каждую пробирку наливают растворы питательной среды в следующих количествах:
|
Растворы |
Количество, мл |
|
20%-ный раствор сахарозы |
1 |
|
Раствор А |
0,5 |
|
Раствор В |
0,5 |
|
ZnSO |
0,4 |
|
CuSО |
0,1 |
|
Буферный раствор |
0,1 |
|
Дистиллированная вода стерильная |
2,0 |
Все пробирки помещают в кипящую водяную баню на 1 мин, затем охлаждают и добавляют в каждую следующие количества растворов витаминов (табл.7).
Таблица 7
|
Витамины |
Количество витаминов, добавленное в каждую пробирку |
|||||||
|
мл |
мкг |
|||||||
|
Аневрин В |
0,1 |
2 |
||||||
|
Рибофлавин В |
0,1 |
1 |
||||||
|
Никотиновая кислота |
0,1 |
50 |
||||||
|
|
0,1 |
3 |
||||||
|
Пиридоксин В |
0,1 |
10 |
||||||
|
Пантотенат кальция |
0,1 |
5 |
||||||
|
Мезоинозитол |
0,1 |
500 |
||||||
|
В восемь пробирок из девяти добавляют раствор чистого биотина в возрастающем количестве. |
||||||||
|
Номер пробирки |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Количество биотина, мкг |
0,001 |
0,003 |
0,005 |
0,0075 |
0,01 |
0,02 |
0,03 |
0,04 |
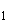
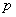 -Аминобензойная кислота
-Аминобензойная кислотаЗатем во все девять пробирок вносят эмульсию дрожжей с таким расчетом, чтобы в каждой пробирке было одинаковое количество дрожжевых клеток - около 1 млн. Для этого одну петлю агаровой культуры размешивают в 10 мл стерильного физиологического раствора и определяют количество клеток в 1 мл эмульсии путем подсчета в счетной камере или по нефелометру.
После внесения дрожжей содержимое пробирок доводят до метки стерильной дистиллированной водой и переливают в стерильные колбочки. Пробирки ополаскивают 1 мл той же воды так, чтобы в колбочках получилось всего 11 мл жидкости в каждой. Приготовление стандартного ряда и засев колбочек производят в боксе или в отдельной чистой комнате без применения горелки.
Засеянные дрожжами колбочки помещают в термостат при 30 °С на 20-24 ч. В течение этого времени их встряхивают 3-4 раза и поддерживают рН среды в пределах 4,5-5,5, добавляя в случае надобности гидроксид калия. По окончании срока выращивания подсчитывают количество клеток выросших дрожжей при помощи счетной камеры или нефелометра.
По мере увеличения дозы биотина повышается количество выросших клеток в среде. Результаты подсчета приведены в табл.8.
Таблица 8
|
Номер колбы |
Количество добавленного биотина, мкг |
Количество выросших клеток |
|
1 |
0,001 |
11,17 |
|
2 |
0,003 |
22,7 |
|
3 |
0,005 |
27,6 |
|
4 |
0,0075 |
40,0 |
|
5 |
0,01 |
45,0 |
|
6 |
0,02 |
47,0 |
|
7 |
0,03 |
47,0 |
|
8 |
0,04 |
47,0 |
|
9 |
Контроль без биотина |
2-2,5 |
По данным, приведенным в табл.8, вычерчивают стандартную кривую, которой и пользуются при определении биотина.
МЕТОДЫ ПОСТРОЕНИЯ ГРАДУИРОВОЧНЫХ ТАБЛИЦ И ГРАФИКОВ (ГРАДУИРОВКА ПРИБОРА ФЭК-56)
В зависимости от назначения прибора изготовляют соответствующие градуировочные таблицы и графики.
Для определения количества прессованных дрожжей (с 75%-ной влажностью) в единице объема, например грамме на литр, изготовляют ряд взвесей дрожжей: навески прессованных дрожжей с 75%-ной влажностью 10, 15, 17, 20 и т.д. до 70-80 г размешивают в 1 л воды. Заготовленные взвеси дополнительно разбавляют водой (водопроводной) в соотношении 1:50; 3:50, так как оптическая плотность исходных взвесей слишком велика и недоступна определению на приборе.
Взвеси подвергают измерениям и на основании полученных цифр составляют таблицу и строят калибровочный график, откладывая по горизонтальной оси известные концентрации, а по вертикальной - соответствующие им значения оптической плотности. Калибровочный график и таблицу следует время от времени проверять.
Для составления градуировочной таблицы по количеству клеток дрожжей в 1 мл взвеси следует пользоваться счетной камерой. Приготовляют ряд взвесей дрожжей. Взвеси готовят с таким расчетом, чтобы в них содержалось от 1,5 до 40 млн. клеток в 1 мл. При большем количестве клеток снижается точность определения (слишком большая оптическая плотность).
Во взвесях подсчитывают количество клеток в 1 мл и параллельно определяют оптическую плотность их. На основании полученных данных составляют таблицу и график.
Используя прибор ФЭК-56 для количественного определения содержания в продукте активаторов роста дрожжей, например биотина, так как имеется определенная зависимость между количеством биотина в среде и количеством выросших клеток дрожжей, а следовательно, и оптической плотностью, градуировочную таблицу и далее кривую составляют по трем показателям: количеству добавленного биотина; количеству выросших клеток; изменению оптической плотности по каждой из определяемых точек.
В дальнейшем градуировочную таблицу и кривую строят только по содержанию чистого биотина в среде и соответственному изменению оптической плотности.
|
Показания нефелометра |
0,395 |
0,450 |
0,520 |
0,535 |
0,550 |
0,555 |
0,555 |
0,540 |
|
Количество биотина в среде, мкг |
0,001 |
0,003 |
0,005 |
0,0075 |
0,01 |
0,02 |
0,02 |
0,04 |
ТЕХНОХИМИЧЕСКИЙ КОНТРОЛЬ ПРОИЗВОДСТВА
АНАЛИЗ МЕЛАССЫ
Меласса, предназначенная для выработки хлебопекарных дрожжей, должна удовлетворять следующим требованиям: консистенция однородная, густая, сиропообразная; цвет темно-бурый; запах, присущий свекловичной мелассе, без постороннего запаха.
Меласса должна легко и быстро растворяться в воде в любых соотношениях без образования осадка. Меласса, отвечающая требованиям дрожжевого производства, должна иметь следующие показатели:
|
Сухие вещества, %, не менее |
74 |
|
Сахар (по прямой поляризации), % |
От 46 до 50 |
|
Инвертный сахар, %, не более |
2,0 |
|
Доброкачественность, % |
|
|
не более |
65 |
|
не менее |
55 |
|
рН |
6,5-8,5 |
|
Общий азот, %, не менее |
1,4 |
|
Аминный азот, %, не менее |
0,3 |
|
Зола (за вычетом кальция), %, не менее |
7,0 |
|
Калий (K |
3,5 |
|
Кальций (СаО), %, не более |
1,0 |
|
Биотин, мкг/кг |
200 |
|
Цветность, мл 0,1 н. раствора йода на 100 мл 2%-ного раствора мелассы, не более |
2,0 |
|
Сернистый ангидрид, %, не более |
0,05 |
|
Летучие кислоты, %, не более |
1,2 |
Отбор проб
Пробы мелассы для анализа отбираются представителем отдела сырья, работником ОТК или работником заводской лаборатории.
Пробу отбирают в чистую сухую посуду из каждой прибывшей цистерны во время слива. Если слив производится без подогрева, то отбор осуществляется через краник, установленный на корыте для слива мелассы, непрерывно в течение всего времени опорожнения цистерны. В случае необходимости подогрева пробу отбирают через верхний люк цистерны пробоотборником из трех слоев: верхнего, среднего и нижнего.
Масса пробы составляет не менее 1,5 кг от каждых 10 т мелассы при приеме по массе и не менее 10 кг при приеме по объему.
В случае, если цистерны поступили от одного завода-отправителя одновременно, отобранные из цистерн пробы смешивают в одной емкости чистой сухой деревянной лопаткой, не допуская образования пены.
Из средней пробы отбирают в три чистые сухие бутылки по 0,5 л. Одну передают в лабораторию завода для анализа, две другие опечатывают печатью завода или пломбируют. На бутылки наклеивают этикетки с указанием наименования продукта, завода-отправителя, завода-получателя, номер цистерны, номер железнодорожной накладной, даты отбора пробы. Пробы хранят в течение 2 мес, а в случае разногласий - до окончания спора.
Определение концентрации сухих веществ
Рефрактометрический метод. Рефрактометр применяется при контроле дрожжевого производства для определения сухих веществ в мелассе и мелассных растворах. Он представляет собой прибор для измерения величины преломления луча света различными веществами. Рефракция света наступает, когда пучок параллельных лучей, направленных наклонно к плоскости раздела двух сред, переходит из одной среды в другую, причем скорость распространения света в этих средах неодинакова.
Испытуемую жидкость помещают между двумя призмами рефрактометра, коэффициент преломления которых больше, чем испытуемой жидкости, и граница света и тени, определяемая величиной угла преломления, падает на шкалу, где нанесены величины преломления, соответствующие данной величине угла. Зная показатель преломления одной среды, можно вычислить показатель преломления другой.
На рефрактометрах, применяемых в лабораторной практике, на одной шкале отсчитывают непосредственно коэффициент преломления, а на другой - соответствующее содержание сухих веществ в среде.
Лабораторный рефрактометр (рис.8) широко применяют на дрожжевых заводах. Он состоит из основания 11 и стойки 10 . Все оптические части рефрактометра помещены в закрытый металлический корпус 1 . Снаружи находятся только поверхности призм 2 и 3 и окошко, внутри которого имеются шкала 4 и окуляр 6 . Призмы заключены в металлическую оправу, верхняя из них укреплена на шарнире, ее можно откидывать вверх при помощи ручки.
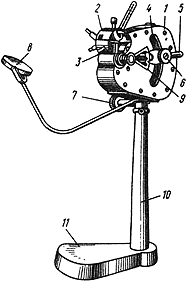
Рис.8. Лабораторный рефрактометр:
1 - корпус прибора; 2 - осветительная призма; 3 - измерительная призма; 4 - шкала; 5 - ручка для перевода нониуса поля; 6 - окуляр поля зрения; 7 - винт, укрепляющий положение зеркала; 8 - осветительное зеркало; 9 - секторный рычаг компенсатора; 10 - стойка; 11 - основание
На призмы направляют свет (при помощи зеркала 8 ). Окуляр 6 можно опускать и поднимать, так как он укреплен на рычаге, поворачивающемся вокруг оси. Поворотом головки окуляр 6 устанавливают на фокус, при этом с правой стороны поля зрения видна шкала с делениями от 0 до 95%, из них одна часть от 0 до 50% имеет деления по 0,2%, а другая от 50 до 95% - по 0,1%. С левой стороны расположена шкала показателей преломления от 1,3 до 1,54.
Показание рефрактометра устанавливают по пунктирной линии. Поднимая и опуская рычаг окуляра, наводят эту линию на границу раздела темного и светлого полей зрения и отмечают на шкалах соответствующие показания.
Содержание сухих веществ при помощи рефрактометра определяют при температуре 20 °С. Для этого призмы рефрактометра включены в специальную оправу, в которой циркулирует вода указанной температуры. Если при снятии показаний рефрактометра температура исследуемого раствора имела отклонение от стандартной (20 °С), то в полученное значение необходимо внести температурную поправку. Если температура при определении сухих веществ была ниже 20 °С, то величину поправки вычитают из найденного количества. Если температура при определении выше 20 °С, то величину поправки следует прибавить к найденному количеству сухих веществ (см. табл.1 приложения IV).
Для определения сухих веществ рефрактометром 2-3 капли мелассы, не разбавленной или разбавленной водой в соотношении 1:1, наносят на центральную часть нижней призмы рефрактометра при помощи оплавленной стеклянной палочки. Верхнюю призму отпускают и плотно прижимают к нижней неподвижной призме. Затем путем передвижения окуляра находят наиболее резкую границу между темной и светлой половиной поля зрения и отмечают по шкале процентное содержание сухих веществ.
Содержание сухих веществ в мелассе (разбавленной водой в соотношении 1:1), выраженное в процентах, равно удвоенному показанию рефрактометра.
Для проверки правильности показания прибора на призму наносят каплю чистой дистиллированной воды и наблюдают за показаниями рефрактометра. Если пунктирная линия или центр кружка отклоняются от нуля при повторных определениях более чем на 0,2 показания правой шкалы, то при помощи специального ключа устанавливают ее нулевое положение.
При применении рефрактометра содержание сухих веществ в мелассных растворах с разбавлением исходной мелассы 1:1 увеличивается на 3-5% по сравнению с тем же показателем, полученным при определении сухих веществ в той же мелассе сахарометром.
В случае отсутствия рефрактометра или его неисправности сухие вещества определяют при помощи сахарометра.
Определение содержания сухих веществ сахарометром. Определение производят в растворе мелассы, разбавленной водой в соотношении 1:1. На технических весах тарируют стакан со стеклянной палочкой и взвешивают в нем 100 г исследуемой мелассы. К массе мелассы добавляют 60-70 мл горячей дистиллированной воды и тщательно перемешивают. Смесь охлаждают до 20 °С, доводят дистиллированной водой (20 °С) до двойной массы мелассы (200 г), снова перемешивают и определяют в растворе содержание сухих веществ сахарометром.
Если при определении температура раствора имела отклонение от стандартной (20 °С), то в полученное значение необходимо ввести температурную поправку (см. табл.2 приложения IV).
Содержание сухих веществ в мелассе, выраженное в процентах, равно удвоенному показанию сахарометра.
Определение состава сахаров
В контроле дрожжевого производства точное определение содержания сахаров имеет большое значение, так как по количеству сахара в сырье определяют выход дрожжей. При поступлении мелассы на завод производят полный анализ состава сахаров и определение суммы сбраживаемых сахаров.
Для количественного определения содержания сахаров в мелассе служат поляриметрические и химические методы. Прямая поляризация не отражает истинной сахаристости мелассы, и общее содержание сбраживаемых сахаров в ней определяют по прямой и инверсионной поляризации. Кроме того, инвертный сахар определяют химическим методом (по Офнеру).
Определение содержания сахарозы методом прямой поляризации. Поляриметрический метод определения сахарозы основан на способности углеводов вращать плоскость поляризации луча света, а также на свойстве органических веществ, в молекуле которых имеется асимметрический азот углерода, вращать плоскость поляризационного луча. Известно, что колебания обыкновенного светового луча происходят во всех плоскостях, перпендикулярных его направлению.
При прохождении обыкновенного луча света через некоторые кристаллы их решетки способны пропускать лучи только определенного направления колебания. По выходе из кристалла колебания луча будут происходить только в одной плоскости, такой луч называется поляризованным.
Ход определения заключается в следующем. Две с половиной навески мелассы (65 г) при помощи теплой дистиллированной воды переводят без потерь в колбу вместимостью 250 мл, где вместе с ополосками должно быть около 150 мл.
Полученный раствор охлаждают до 20 °С и осветляют раствором азотнокислого свинца (340 г в 1 л) и гидроокиси натрия (32 г в 1 л).
Осветлители сохраняются отдельно. На осветление раствора мелассы расходуется 30-50 мл каждого из растворов. Для лучшего осветления и большей точности результатов анализа осветляющие реактивы следует вводить по частям (в 4-6 приемов).
Процесс осветления осуществляется следующим образом. В колбу с раствором мелассы прибавляют 5-10 мл раствора азотнокислого свинца и через 15-20 с 5-10 мл раствора гидроокиси натрия. Смесь перемешивают легким вращением колбы в течение 15-20 с. Затем опять в указанном порядке наливают осветлители. Так повторяют несколько раз в зависимости от полноты осветления. Содержимое колбы доводят дистиллированной водой почти до метки, удаляют пену каплей эфира и доливают водой точно до метки при температуре 20 °С. Приставшие к шейке колбы капельки воды вытирают фильтровальной бумагой, после чего взбалтывают содержимое колбы и фильтруют через сухой бумажный фильтр, вставленный в сухую воронку, которую покрывают сверху часовым стеклом. Фильтрат собирают в сухую коническую колбу. Первые капли фильтрата выливают. Если фильтрат мутный, то его возвращают на фильтр до тех пор, пока не получится совершенно прозрачный раствор.
В целях удаления избытка свинца на каждые 100 мл полученного фильтрата в колбу прибавляют 0,3 г сухого измельченного порошка однозамещенного фосфата аммония (NННРО). Для его растворения жидкость энергично взбалтывают, после чего прибавляют на каждые 100 мл фильтрата по 0,2 г гидросульфита натрия (NaSO) или бисульфита натрия (NaHSO). Затем раствор перемешивают и оставляют на 20 мин, снова взбалтывают и фильтруют согласно предыдущим указаниям.
Полученный раствор является исходным для определения прямой и инверсионной поляризаций, а также инвертного сахара.
Осветленный раствор наливают в трубку высотой 200 мм и поляризуют в сахариметре при температуре 20 °С. Для каждой пробы проводят не менее трех отсчетов и принимают средний. Полученная величина и есть прямая поляризация раствора, обозначаемая буквой 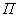 .
.
Поляриметрические исследования ведут при помощи поляриметров. В дрожжевом производстве в настоящее время получил широкое распространение поляриметр СУ-3. Ниже приведена его оптическая схема.
Оптическая схема прибора построена следующим образом (рис.9). Свет от электролампочки 1 проходит через матовое стекло 3 , предназначенное для рассеивания света (вместо матового стекла в оптическую систему может быть введен светофильтр 2 ). Далее световой поток проходит конденсорную линзу 4 , попадает на поляризатор 5 и выходит из него линейно-поляризованным. За поляризатором находятся два защитных стекла 6 и 7 , между которыми устанавливается поляриметрическая кювета с исследуемым раствором. Подвижной кварцевый клин 8 , стеклянный контрклин 9 и неподвижный кварцевый клин 10 образуют кварцевый компенсатор, который компенсирует вращение плоскости поляризации, вызванное исследуемым раствором. За кварцевым компенсатором установлен анализатор 11 , обнаруживающий поворот плоскости поляризации. Зрительная труба, состоящая из двухлинзового объектива 12 и окуляра 13 , сфокусирована на выходную грань поляризатора 5 . При помощи зрительной трубы наблюдают в увеличенном виде линию раздела поляризатора.
Рис. 9. Оптическая схема поляриметра СУ-3
Свет от электролампочки служит также для освещения шкалы 16 и нониуса 17 , что достигается при помощи отражательной призмы 14 и защитного стекла 15 , рассеивающего свет. Цифры и деления шкалы 16 и нониуса 17 рассматриваются в увеличенном виде при помощи лупы, состоящей из двух линз 18 .
Шкала связана с подвижным кварцевым клином. Таким образом, смещение подвижного кварцевого клина пропорционально углу вращения плоскости поляризации, вызванное исследуемым раствором, передается на шкалу и отсчитывается при помощи лупы.
Устройство прибора. Основными частями прибора (рис.10) являются узел измерительной головки 1 и осветительный узел 2 , соединенные между собой траверсой 3 , на которой укреплена камера 4 для поляриметрических трубок.
Рис.10. Общий вид поляризатора СУ-3
В передней части прибора имеется лупа 5 в оправе для отсчета показаний по шкале и зрительная труба 6 , в которой находится гильза 7 . С тыльной стороны измерительной головки находится узел нониуса. В нижней части измерительной головки расположена рукоятка кремальерной передачи 8 для перемещения подвижного кварцевого клина и связанной с ним шкалы.
Осветительный узел имеет поворотную обойму 9 со стеклянным светофильтром и матовым стеклом и патрон с лампочкой 10 .
Траверса крепится болтом к литому основанию 11 с вмонтированным внутрь его трансформатором, через который питается лампочка от сети переменного тока 220 В.
Калибровка шкалы поляриметра. Условную шкалу поляриметра калибруют по раствору химически чистой сахарозы. При поляризации раствора, содержащего точно 26 г сахарозы в 100 мл, в 200-миллиметровой трубке получают показание 100° условной шкалы. Таким образом, одно деление шкалы соответствует 0,26 г сахарозы. Шкала поляриметра при определении сахара показывает процентное содержание его. Для отсчета десятых долей предназначен нониус.
Перед началом работы необходимо установить прибор на нуль. При этом нулевое значение шкалы должно совпадать с нулем нониуса (рис.11,). После этого трубку заполняют исследуемым раствором, помещают в камеру прибора и производят отсчет показаний шкалы и нониуса. На рис.11, показано положение шкалы относительно нониуса, соответствующее отсчету +24,5°. При этом нуль нониуса расположен после 24 полных делений шкалы, а пятое деление нониуса совпало с одним из делений шкалы.
Рис.11. Отсчет показаний на шкале поляриметра
При анализе левовращающихся растворов все операции проводят в обратном направлении, в этом случае нуль нониуса расположен левее шкалы.
Поляризационная трубка длиной 200 мм считается нормальной. При поляризации в трубках длиной 100 или 400 мм показания поляриметра в первом случае удваивают, а во втором уменьшают в 2 раза.
Для трубок длиной 100 и 200 мм допускаются отклонения ±0,05 мм, для трубок длиной 400 мм - ±0,1 мм. Края трубок должны быть хорошо пришлифованы, а внутренняя поверхность - зеркально чистой. При наличии на ней налета, не удаляемого водой, необходимо промывать трубки слабым раствором уксусной кислоты, а затем водой. Поляризационную трубку закрывают с обоих концов хорошо пришлифованными и чистыми покровными стеклами и завинчивают гайками с эластичной резиновой прокладкой.
Трубку наполняют следующим образом. Один конец ее закрывают пришлифованным круглым стеклом и завинчивают гайкой. Затем, слегка наклонив трубку, заполняют ее испытуемым раствором так, чтобы жидкость выступала над краем трубки при вертикальном положении. Осторожно, срезая жидкость стеклом, закрывают и затем завинчивают трубку. В жидкости не должно оставаться пузырьков воздуха. Протерев стекла, производят поляризацию. Температура поляризуемого раствора должна быть равна 20 °С (в течение всего периода поляризации), поэтому поляриметр устанавливают в комнате, имеющей постоянную температуру 20 °С, или в футляре, входное отверстие которого завешивают шторой из черной ткани.
Прямую поляризацию осветленного раствора проводят в трубке длиной 200 мм при температуре 20 °С.
Для каждой пробы производят не менее трех отсчетов по поляриметру и принимают средний. Полученная величина и есть прямая поляризация раствора, обозначенная буквой .
Определение инверсионной поляризации. Для определения суммы сбраживаемых сахаров в мелассе производят инверсионную поляризацию, используя фильтрат, полученный для прямой поляризации.
Ход определения заключается в следующем. Отбирают 50 мл фильтрата (полученного для определения сахарозы по прямой поляризации), соответствующие 13 г мелассы, переводят их в колбу вместимостью 100 мл, приливают туда же 30 мл раствора соляной кислоты (одна часть соляной кислоты относительной плотности 1,19 и пять частей воды по объему), вращательным движением колбы перемешивают содержимое, опускают в раствор термометр и колбу помещают в водяную баню, предварительно нагретую до температуры 75 °С.
Колбу устанавливают не на дно бани, а на фарфоровую или металлическую вставку с круглыми вырезами.
Температуру воды в бане поддерживают в пределах 70-72 °С, нагревая жидкость в колбе в течение 2,5-3 мин до 67-69 °С и поддерживая эту температуру в течение 5 мин. После этого колбу из бани вынимают и быстро (не более 2,5 мин) охлаждают под краном до 20 °С. Весь процесс инверсии должен быть проведен в течение 10 мин.
После охлаждения раствора термометр вынимают и ополаскивают водой, доводят содержимое колбы до метки, тщательно взбалтывают, фильтруют и поляризуют в трубке длиной 200 мм точно при 20 °С. Удвоенное показание поляриметра дает инверсионную поляризацию, обозначаемую буквой 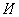 . В случае отклонения температуры при поляризации раствора от 20 °С следует вносить поправку на температуру по формуле
. В случае отклонения температуры при поляризации раствора от 20 °С следует вносить поправку на температуру по формуле
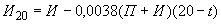 ,
,
где и - инверсионная и прямая поляризация, %; - температура, °С.
Определение содержания инвертного сахара методом Офнера. Из осветленного раствора, оставшегося после определения прямой поляризации, отбирают пипеткой 10 мл (соответствующее 2,6 г мелассы), переводят в мерную колбу на 100 мл и доливают дистиллированной водой до метки. Отбирают пипеткой 25 мл разбавленного таким образом раствора (соответствующего 0,65 г мелассы), переводят в коническую колбу вместимостью 150 мл и прибавляют 25 мл реактива Офнера.
В смешанный раствор добавляют на кончике ножа немного талька в порошке или грубо измельченной пемзы. Колбу ставят на асбестовую сетку с вырезанными в центре отверстиями диаметром около 6,5 см и нагревают на газовой горелке или спиртовке в течение 4-5 мин до начала кипения, затем уменьшают пламя так, чтобы оно едва касалось сетки, и поддерживают умеренное кипение точно 7 мин, по истечении которых содержимое колбы охлаждают до 20 °С в холодной воде, не взбалтывая во избежание окисления осадка. При нагреве на электрической плитке кипение регулируют, включая и выключая плитку.
7,5 мл 1 н. раствора соляной кислоты, отмеренного градуированным цилиндром, осторожно прибавляют по стенкам колбы в охлажденный раствор, чтобы растворить находящийся на стенках осадок, и сразу же после добавления кислоты прибавляют к испытуемому раствору из бюретки 20 мл 0,0323 н. раствора йода.
Колбу закрывают стеклянной или корковой пробкой и оставляют на 2 мин, периодически перемешивая содержимое колбы вращением. Ровно через 2 мин оттитровывают избыток йода в колбе 0,0323 н. раствором гипосульфита. В конце титрования, когда раствор станет светло-желтым, в него добавляют 2,5 мл 0,5%-ного раствора крахмала и титруют до перехода окраски от синей в зеленую или бронзовую.
Одновременно проводят контрольный опыт (с тем же количеством раствора мелассы, реактивом йода, что и в основном опыте, но без кипячения) для установления поправки на окисляемость йодом веществ, находящихся в растворе.
По разности между количеством миллилитров гипосульфита, пошедшим на титрование в рабочем и контрольном опытах, устанавливают количество связавшегося йода. Содержание инвертного сахара определяют из расчета, что 1 мл 0,0323 н. раствора йода эквивалентен 1 мг инвертного сахара, по формуле
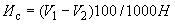 ,
,
где 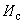 - содержание инвертного сахара к массе мелассы, %; , - соответственно объем 0,0323 н. раствора серноватистокислого натрия в контрольном и рабочем опыте, мл; - навеска мелассы, г.
- содержание инвертного сахара к массе мелассы, %; , - соответственно объем 0,0323 н. раствора серноватистокислого натрия в контрольном и рабочем опыте, мл; - навеска мелассы, г.
Пример. На определение инвертного сахара к 25 мл раствора, полученного разведением в 10 раз (10:100) раствора, подготовленного для поляризации (соответствующего 0,65 г мелассы), прибавили 20 мл 0,0323 н. раствора йода. На титрование избытка йода затратили 14,6 мл 0,0323 н. раствора гипосульфита. На то же количество йода в контрольном опыте израсходовали 19,4 мл гипосульфита. Следовательно, количество йода, вступившего в реакцию, будет равно 4,8 мл (19,4-14,6), т.е. в 0,65 г мелассы содержится 4,8 мг инвертного сахара, или в процентах к массе
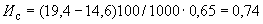 %.
%.
Расчет сахаристости (суммы сбраживаемых сахаров). Сахаристость рассчитывают по формуле
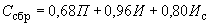 ,
,
где  - сахаристость мелассы, %; и - абсолютные значения прямой и инверсионной поляризаций, %; - содержание инвертного сахара, определенное химическим методом (по Офнеру).
- сахаристость мелассы, %; и - абсолютные значения прямой и инверсионной поляризаций, %; - содержание инвертного сахара, определенное химическим методом (по Офнеру).
Пример.  %;
%; 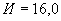 %;
%; 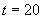 °С;
°С; 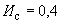 %.
%.
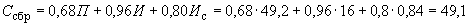 %.
%.
Для более полной оценки качества мелассы, поступившей на дрожжевые заводы, определяют содержание сахарозы и рафинозы в ней по формулам:
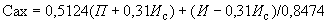 ;
;
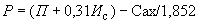 ,
,
где 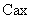 и
и 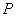 - содержание сахарозы и рафинозы в мелассе, %; и - абсолютные значения прямой и инверсионной поляризаций;
- содержание сахарозы и рафинозы в мелассе, %; и - абсолютные значения прямой и инверсионной поляризаций;  - поправки на вращательную способность инвертного сахара , определяемого химическим методом.
- поправки на вращательную способность инвертного сахара , определяемого химическим методом.
Пример. %; %; °С;  %.
%.
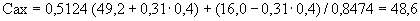 %.
%.
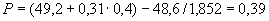 %.
%.
Определение доброкачественности мелассы
Доброкачественность - это отношение сахара к общему количеству сухих веществ. Она рассчитывается по формуле
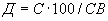 ,
,
где 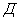 - доброкачественность мелассы, %; - содержание сахара в мелассе, % по прямой поляризации (если сумма сбраживаемых сахаров превышает количество сахара, полученное по прямой поляризации, более чем на 1%, то содержание сахара в мелассе считают по сумме сбраживаемых сахаров);
- доброкачественность мелассы, %; - содержание сахара в мелассе, % по прямой поляризации (если сумма сбраживаемых сахаров превышает количество сахара, полученное по прямой поляризации, более чем на 1%, то содержание сахара в мелассе считают по сумме сбраживаемых сахаров);  - содержание сухих веществ в мелассе, %.
- содержание сухих веществ в мелассе, %.
Пример. =48%, =80%. Доброкачественность мелассы составит
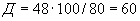 %.
%.
Доброкачественность мелассы, используемой в дрожжевом производстве, составляет 55-65%, т.е. на 100 массовых частей сухого вещества мелассы должно приходиться 55-65 массовых частей сахара. Хорошо перерабатывается меласса с невысокой доброкачественностью (58-60%).
При доброкачественности более 65% в мелассе имеется избыток сахара и недостаток несахаров, что препятствует активному размножению дрожжей. При доброкачественности ниже 55% в мелассе отмечаются, как правило, низкое содержание сахара (39-44%) и пониженная активная кислотность (рН 5-6,2). Такая меласса плохо хранится, и в ней могут протекать различные микробиологические и биохимические процессы.
Быстрый метод определения содержания мелассы в мелассных растворах при поступлении их в производство
Для быстрого учета сахара в мелассе используют пробы, взятые из сборников, куда поступает меласса после перемешивания (гомогенизации) с добавлением воды, например при разбавлении исходного сырья до плотности 35-17% СВ. При этом пользуются номограммами (рис.12, 13, 14).
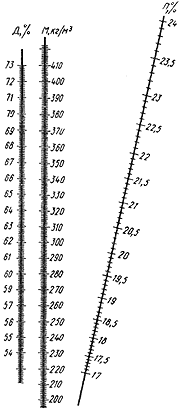
Рис.12. Номограмма для определения количества мелассы (с содержанием сахара 46%) в 1 м раствора при содержании сухих веществ 17-24%
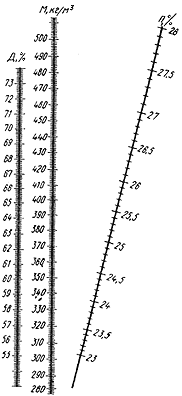
Рис.13. Номограмма для раствора мелассы, содержащей 23-28% сухих веществ
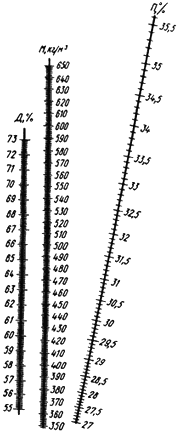
Рис.14. Номограмма для раствора мелассы, содержащей 27-36,5% сухих веществ
Для определения сахара в мелассных растворах требуется учитывать доброкачественность исходного сырья и концентрацию сухих веществ раствора мелассы по сахарометру в пробах, отобранных из сборника.
При помощи линейки соединяют две точки и , указанные на шкалах, и находят величину - количество мелассы (в кг) с содержанием 46% сахара в 1 м.
Определение активной кислотности (рН)
Активную кислотность (рН) мелассы определяют с помощью лабораторных рН-метров со стеклянными электродами. Для определения рН к навеске мелассы (20 г), взятой на технохимических весах, добавляют 20 г дистиллированной воды, тщательно перемешивают и определяют рН раствора обычно при помощи лабораторных рН-метров, строго соблюдая инструкцию по эксплуатации к лабораторному рН-метру. Показания рН-метра записывают по истечении 3 мин после погружения электродов. В настоящее время на дрожжевых заводах применяются лабораторные рН-метры ЛПУ-01 и рН-340.
Пределы измерения величины рН от 1 до 14 с диапазонами: 1-2; 2-5; 5-8; 8-11; 11-14. Погрешность прибора не превышает ±0,6% для диапазона измерения от 1 до 14 и от 0,04 до 0,05% для приведенных выше интервалов.
В приборе рН-340 предусмотрена автоматическая компенсация температуры исследуемого раствора и с помощью термометра.
Температурной компенсацией с помощью термометра рекомендуется пользоваться при измерении рН растворов, имеющих постоянную температуру. При этом ручку на лицевой панели с надписью "Температура раствора" следует установить на значение температуры исследуемого раствора. Для автоматической температурной компенсации применяется специальный термокомпенсатор, входящий в комплект датчика ДЛ-02.
Лабораторный датчик ДЛ-02 предназначен для крепления электродов и установки сосуда с исследуемым раствором при измерении рН. Датчик рассчитан на применение измерительных стеклянных электродов типа ЭСЛ и проточного хлорсеребряного электрода сравнения.
Все элементы датчика собраны на настольном вертикальном штативе, в верхней части которого установлен проточный, вспомогательный электрод сравнения.
Для измерения величины рН используется система со стеклянным и вспомогательным электродом, схема которой приведена на рис.15.
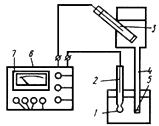
Рис.15. Схема измерения величины рН раствора с помощью рН-метра-милливольтметра рН-340
Сущность метода: при погружении стеклянного электрода 1 в исследуемый раствор между поверхностью шарика электрода и раствором происходит обмен ионами, в результате которого ионы лития в поверхностных слоях стекла замещаются ионами водорода и стеклянный электрод приобретает свойства водородного электрода.
Между поверхностью стекла и исследуемым раствором возникает разность потенциалов, величина которой определяется активностью ионов водорода в растворе.
Для создания электрической цепи при измерении применяются контактные электроды: внутренний контактный электрод 2 , заполняющий внутреннюю часть стеклянного электрода, и внешний контактный электрод 3 (вспомогательный), осуществляющий контакт с исследуемым раствором.
Вспомогательный хлорсеребряный электрод соединен с исследуемым раствором при помощи электролитического ключа 4 трубки, наполненной раствором хлористого калия, заканчивающейся пористой перегородкой 5 , через которую непрерывно просачивается раствор хлористого калия, предотвращая проникновение из исследуемого раствора в систему электрода 3 посторонних ионов, которые могли бы изменить величину электродвижущейся силы электрода.
Перед определением рН стеклянный электрод "вымачивают" в 0,1 н. растворе соляной кислоты в течение 8 ч, после чего электрод необходимо тщательно промыть дистиллированной водой и удалить избыток воды фильтровальной бумагой.
Отсчет ведется по шкале 7 прибора 6 в зависимости от рН исследуемого раствора. Показания при диапазоне измерения от 1 до 14 снимают по нижней шкале через 1-3 мин после погружения электрода. При анализе раствора, рН которого находится в узких пределах, например 2-5, отсчет ведут по соответствующей шкале.
По окончании работы с прибором электроды следует оставить погруженными в воду. Для обеспечения правильной работы рН-метра необходимо периодически проводить проверку прибора по стандартным буферным растворам. Рекомендуется применять буферный раствор, рН которого находится в том же диапазоне измерения, что и рН исследуемых растворов.
Буферные растворы приготовляют из реактивов для рН-метрии. Эти реактивы выпускаются в виде фиксаналов. При эксплуатации прибора не следует допускать высыхания стеклянного электрода, так как это может привести к изменению его характеристик. Вспомогательный электрод необходимо периодически пополнять насыщенным при комнатной температуре раствором хлористого калия.
Определение содержания азота в мелассе
Азотные вещества мелассы в процессе выращивания дрожжей подвергаются изменениям с образованием новых соединений и в значительной мере определяют выход и качество готовой продукции.
Определение содержания общего азота. Общий азот в мелассе определяют по методу Кьельдаля, основанному на окислении органических веществ концентрированной серной кислотой при нагревании. Содержание общего азота в мелассе может колебаться в пределах от 0,8 до 2,2%.
Для определения азота по Кьельдалю берут от 1,5 до 2,5 г мелассы. Навеску отвешивают на аналитических весах в пробирке, которую вводят в колбу Кьельдаля, и выливают содержимое. При этом наблюдают, чтобы частицы испытуемого вещества не прилипали к горлышку колбы. Массу мелассы, взятую для анализа, определяют по разности между массой пробирки с навеской и массой пробирки после выливания мелассы. В эту же колбу с мелассой наливают 20-30 мл концентрированной химически чистой серной кислоты (относительной плотностью 1,84) и дают кислоте хорошо пропитать мелассу в течение 2-4 ч (лучше 8-10 ч), чтобы избежать пенения в процессе сжигания.
Для ускорения сжигания в колбу добавляют катализаторы. Наилучшим катализатором при сжигании органических веществ в серной кислоте является селен, которого достаточно добавить несколько крупинок. В качестве катализаторов применяют также сернокислый калий (по 1-1,5 г на каждые 10 мл серной кислоты) и сульфат меди (0,5-1 г).
Горло колбы закрывают специальной стеклянной пробкой с расширением, неплотно закрывающей колбу. Для кипячения колбу ставят в наклонное положение на сетке, чтобы ее длинное горло не нагревалось. Это горло служит воздушным холодильником; кипячение кислоты не должно быть слишком бурным, чтобы ее пары успевали сконденсироваться в горле колбы.
После того как жидкость перестанет пениться и выделение белых паров сернистого газа станет более равномерным, нагревание усиливают. Обугленные частички смывают со стенок колбы содержащейся в ней жидкостью, которая к концу реакции обесцвечивается или при сжигании с сульфатом меди приобретает голубую окраску. После полного просветления жидкости нагревание продолжают еще в течение 1-2 ч, при этом избегают бурного кипения ее и следят за тем, чтобы горло колбы не нагревалось сильно. После окончания сжигания навески в содержимом колбы определяют количество аммиака, для этого его перегоняют в колбу с титрованным раствором серной кислоты.
Перегонку производят или в специальном приборе, где можно одновременно проводить несколько определений, или в аппарате, который собирают следующим образом. Круглодонную или плоскодонную колбу вместимостью 1 л устанавливают на штатив, соединяют ее плотной резиновой пробкой с каплеуловителем, а затем с холодильником. В пробку, кроме трубки каплеуловителя, вставляют стеклянную трубку, соединенную каучуковой трубкой с небольшой воронкой, диаметром 4-5 см; на каучуковой трубке имеется винтовой зажим (рис.16).
Рис.16. Прибор для определения азота по Кьельдалю
К другому концу холодильника присоединяют стеклянную трубку с шарообразным расширением в верхней части, которую опускают почти до дна приемника. Приемником может служить обычная коническая колба вместимостью 500 мл. Все части прибора должны быть плотно пригнаны. В приемник наливают 50 мл 0,1 н. раствора серной кислоты и 1-2 капли раствора метилового красного и устанавливают его так, чтобы конец трубки с шарообразным расширением был погружен в кислоту.
Содержимое колбы Кьельдаля после охлаждения разбавляют дистиллированной водой небольшими порциями и сливают через воронку в колбу для отгонки. Колбу Кьельдаля ополаскивают несколько раз водой с таким расчетом, чтобы общий объем жидкости составлял около 400-500 мл. Колбу помещают на штатив и собирают аппарат, как было указано выше. Затем открывают винтовой зажим воронки и наливают 50-100 мл 33%-ного раствора гидроксида натрия. Воронку смывают небольшим количеством дистиллированной воды, завинчивают зажим и содержимое колбы взбалтывают. Наличие в колбе для отгонки избытка щелочи устанавливают при помощи кусочка лакмусовой бумажки, брошенной в колбу. Лакмусовая бумажка синеет.
Колбу для отгонки подогревают горелкой и отгоняют аммиак с водяными парами до тех пор, пока капля отгона не перестанет вызывать посинения красной лакмусовой бумажки. Для этого достаточно отогнать из колбы около половины всей жидкости.
По окончании отгонки конец трубки, опущенный в кислоту, ополаскивают водой в приемник и содержимое его титруют 0,1 н. раствором гидроксида натрия. Затем определяют количество миллилитров 0,1 н. раствора гидроксида натрия, израсходованное на титрование серной кислоты, не связавшейся с аммиаком, и производят расчет.
Содержание общего азота вычисляют по формуле
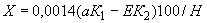 ,
,
где - количество 0,1 н. раствора серной кислоты в приемной колбе, мл; 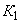 - поправочный коэффициент серной кислоты;
- поправочный коэффициент серной кислоты; 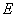 - количество 0,1 н. раствора гидроксида натрия, пошедшее на титрование избытка раствора серной кислоты, мл;
- количество 0,1 н. раствора гидроксида натрия, пошедшее на титрование избытка раствора серной кислоты, мл; 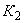 - поправочный коэффициент гидроксида натрия; 0,0014 - количество азота, соответствующее 1 мл 0,1 н. раствора серной кислоты; - навеска мелассы, г.
- поправочный коэффициент гидроксида натрия; 0,0014 - количество азота, соответствующее 1 мл 0,1 н. раствора серной кислоты; - навеска мелассы, г.
Пример. На сжигание взято 2,3716 г мелассы. В приемник налито 50 мл 0,1 н. раствора серной кислоты с поправочным коэффициентом =0,9961. На обратное титрование израсходовано 23,4 мл 0,1 н. раствора гидроксида натрия с поправочным коэффициентом =1,0126. Таким образом, связано аммиаком 50·0,9961-23,4·1,0126=26,2352 мл 0,1 н. раствора серной кислоты.
Содержание общего азота в мелассе составит
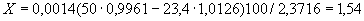 %.
%.
Определение аминного азота "медным способом". Содержание азота аминокислот в мелассе колеблется от 0,1 до 0,5%. Простым и быстрым методом определения количества азота аминокислот является "медный способ". Метод основан на способности аминокислот образовывать комплексные растворимые соединения с медью, содержание которой определяют йодометрическим способом.
Сущность метода заключается в следующем. К определенному количеству испытуемого раствора, содержащего смесь аминокислот, прибавляют при слабощелочной реакции избыток суспензии фосфорнокислой меди в боратно-буферном растворе, в результате чего после взбалтывания в раствор переходят медные соли большинства аминокислот. Затем избыток фосфата меди отфильтровывают и в прозрачном растворе после добавления уксусной кислоты и йодистого калия определяют медь объемным способом по выделившемуся йоду путем титрования гипосульфитом в соответствии с реакцией
 .
.
Каждый миллилитр 0,01 н. раствора NaSO соответствует 0,28 мг аминного азота.
Ход определения заключается в следующем. 13 г мелассы разбавляют дистиллированной водой в мерной колбе на 100 мл. Из полученного раствора отбирают 5 мл в мерную колбочку на 25 мл, добавляют 1 н. раствора едкой щелочи до бледно-синего окрашивания (индикатор тимолфталеин) и 15 мл суспензии фосфорнокислой меди, затем доводят водой содержимое колбы до метки. После фильтрования отбирают 5 мл полученного фильтрата, добавляют 0,25 мл 80%-ной уксусной кислоты и 0,5 йодистого калия (см. приложение II).
Выделившийся йод оттитровывают из микробюретки 0,01 н. раствором гипосульфита (индикатор крахмал). Содержание азота определяют из расчета, что 1 мл 0,01 н. раствора гипосульфита соответствует 0,28 мг аминного азота.
Содержание аминного азота (в %) определяют по формуле
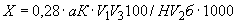 ,
,
где 0,28 - количество аминного азота, эквивалентное 1 мл 0,01 н. раствора гипосульфита, мг; - объем 0,01 н. раствора гипосульфита, пошедший на титрование, мл; - поправочный коэффициент гипосульфита; - навеска мелассы, взятая на анализ, г; - объем разбавленной мелассы, мл; - количество разбавленной мелассы, отобранное на осветление, мл; 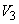 - объем осветленного раствора, мл; - количество фильтрата осветленной мелассы, взятое на определение, мл; 100 - пересчет в проценты; 1000 - перевод миллиграммов в граммы.
- объем осветленного раствора, мл; - количество фильтрата осветленной мелассы, взятое на определение, мл; 100 - пересчет в проценты; 1000 - перевод миллиграммов в граммы.
Обычно на анализ берут 13 г мелассы и проводят одно и то же разбавление ее (=100 мл), отбирают раствор на осветление (=5 мл), доводят объем осветленного раствора до 25 мл (=25 мл) и на определение берут 5 мл фильтрата.
Подставляя эти постоянные величины в уравнение и выполнив сокращения, получим
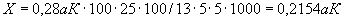 .
.
Пример. На титрование пошло 1,43 мл 0,01 н. раствора гипосульфита (=1,43 мл) с поправочным коэффициентом =0,9875.
Содержание аминного азота в мелассе составит
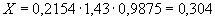 %.
%.
Определение содержания золы в мелассе
В предварительно прокаленную и взвешенную платиновую или фарфоровую чашку (тигель) взвешивают на аналитических весах навеску мелассы около 3 г. Навеску обливают 1-2 мл серной кислоты относительной плотностью 1,84, размешивают платиновой проволочкой или оплавленной стеклянной палочкой, вытирают проволочку или палочку маленьким кусочком фильтровальной бумаги, который также бросают в чашку, и, держа чашку щипцами, осторожно нагревают на небольшом пламени горелки под тягой. Когда меласса перестанет пениться и выделение паров газов прекратится, ее окончательно сжигают и прокаливают до постоянной массы в муфеле при слабом калении или на горелке, при этом доводят платиновую чашку или тигель до слабо-красного каления.
Сжигание навески считается законченным, когда вся масса превратится в белый или розовый порошок, не содержащий черных частиц несгоревшего вещества. Затем чашку или тигель переносят в эксикатор, охлаждают и взвешивают.
Пример. Масса пустого прокаленного тигля 17,9632 г; масса тигля с навеской мелассы 20,9306 г; масса тигля с золой 18,2204 г; навеска мелассы 2,9674 г, масса золы 0,2572 г.
Количество углекислой золы 0,2572·0,9=0,2314 г.
Содержание золы в мелассе 0,2324·100/2,9674=7,8%.
Содержание золы в мелассе колеблется от 6 до 13%.
Определение содержания калия при сухом озолении
В фарфоровую чашку, предварительно прокаленную и доведенную до постоянной массы, берут такое количество веществ, чтобы после озоления навеска золы соответствовала примерно 0,2 г.
К навеске золы приливают в фарфоровую чашку 25-30 мл воды и нагревают на водяной бане 5 мин. Полученный раствор охлаждают, прибавляют 1-2 капли фенолфталеина (1%-ного спиртового раствора) и, приливая по каплям приблизительно полунормальную азотную кислоту, нейтрализуют раствор до обесцвечивания. Если от прибавления фенолфталеина порозовения раствора не наблюдается, то кислоту не прибавляют. Фарфоровую чашку с содержимым снова ставят на водяную баню, нагревают до появления интенсивной окраски и к нагретому раствору приливают по каплям 3-4 капли 0,05 н. раствора азотнокислого серебра для осаждения хлора.
Жидкость перемешивают, фильтруют в мерную колбу на 50 мл и несколько раз промывают малыми количествами горячей воды. К фильтрату прибавляют каплю метилового оранжевого и 0,5 н. раствора азотной кислоты до порозовения раствора (2-3 капли), после чего его охлаждают в струе холодной воды и доводят до метки.
Затем в химический стаканчик вместимостью 50-100 мл пипеткой вносят 5 мл осаждающего раствора Тананаева (реактив 1, см. приложение II). Затем в стаканчик из колбочки приливают пипеткой 25 мл анализируемого раствора. Взяв стаканчик в левую руку и слегка вращая его (для лучшего взаимодействия), правой рукой держат пипетку, из которой приливают испытуемый раствор.
Опорожнив пипетку, содержимое стаканчика перемешивают стеклянной палочкой в течение 2 мин, спустя 5 мин перемешивание ведут еще 1 мин, после чего оставляют содержимое стаканчика на 1-2 ч в покое в темном месте (лучше оставить на ночь). Состав выпавшего осадка комплексной соли калия с кобальтнитритом, содержащим серебро, следующий:
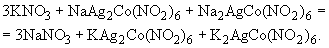
Результаты анализа будут неточными, если в 25 мл исследуемого раствора окажется меньше 3,5 мл или больше 40 мг калия, что должно быть учтено при определении величины навески. Пока осадок отстаивается, тарируют чистый, высушенный при 110 °С стеклянный фильтрующий тигель (ТФ-4-20) и с помощью каучуковой пробки или кольца плотно вставляют его в горло колбы Бунзена, соединенной с насосом.
Одновременно готовят промывалку с дистиллированной водой, подкисленной уксусной кислотой (реактив 6). Спустя 2 ч через тигель сначала сливают жидкость над осадком, а затем переносят на фильтр и сам осадок, взбалтывая его в стаканчике при помощи промывной жидкости, наливаемой из промывалки. При этом частицы осадка, приставшие к стенкам стакана, следует снимать стеклянной палочкой с резиновым наконечником и смывать в тигель. Осадок промывают в тигле до тех пор, пока фильтрат не станет совершенно бесцветным. Достигнув этого, тигель с резиновым кольцом переносят в другую чистую колбу Бунзена и промывают 3-5 мл этилового спирта (реактив 7). Затем тигель снова переносят в чистую колбу Бунзена и промывают осадок 2-3 мл эфира (реактив 8).
Пример. Исходная навеска мелассы равна 1,0386 г. На осаждение калия отобрана половина указанной навески (1,0386 г:2=0,5193 г) мелассы. Масса осадка равна 0,0914 г. Для расчета калия, содержащегося в навеске мелассы, массу осадка умножают на коэффициент, найдя его из табл.9
Массе осадка, равной 0,0914 г, по табл.9 соответствует коэффициент, равный 0,1145. Умножая массу осадка на 0,1145, получаем массу калия в навеске, равную 0,0104 г.
Таблица 9
|
Масса осадка, г |
Коэффициент |
|
0,2800-0,3325 |
0,1196 |
|
0,2270-0,2800 |
0,1186 |
|
0,1735-0,2270 |
0,1167 |
|
0,1300-0,1725 |
0,1155 |
|
0,0875-0,1300 |
0,1145 |
|
0,0660-0,0875 |
0,1138 |
|
0,0446-0,0660 |
0,1228 |
|
0,0220-0,0446 |
0,1124 |
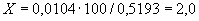 %.
%.
В пересчете на KО это составит 94,2·2,0/78,2=2,4%, где 94,2 - молекулярная масса KO; 78,2 - масса двух атомов калия. Количество KО в золе будет 2,4·100/7,2=33,3%, где 7,2 - содержание золы в мелассе, %.
Ускоренный метод определения калия кобальт-нитритом натрия в азотнокислой среде (ХО ВНИИХП)
На аналитических весах в химическом стаканчике отвешивают 4-5 г мелассы, переводят ее в мерную колбочку вместимостью 100 мл, доводят дистиллированной водой до метки, взбалтывают и фильтруют в сухую колбочку.
В химический стаканчик отбирают пипеткой 10 мл раствора мелассы, добавляют 1,1 мл 1 н. раствора азотной кислоты и 5 мл 20%-ного свежеприготовленного раствора кобальтнитрита натрия, перемешивают палочкой и оставляют стаканчик с палочкой на 2 ч в темном месте (приготовление реактивов см. в приложении II).
Через 2 ч выпавший осадок кобальтнитрита калия фильтруют под разрежением через высушенный при 105 °С и взвешенный стеклянный фильтр ТФ-4-20, установленный в колбе Бунзена, переводя осадок на фильтр 0,01 н. раствором азотной кислоты. После этого осадок на фильтре промывают 10 раз той же кислотой по 2 мл и 5 раз по 2 мл 86-95%-ным этанолом, отсасывают почти досуха, вытирают снаружи фильтр и высушивают в течение часа при 105-110 °С. Фильтр с осадком охлаждают в эксикаторе и взвешивают на аналитических весах. Так как в осадке кобальтнитрита калия калий составляет 17,22%, для расчета содержания его полученную массу осадка умножают на 0,1722 и пересчитывают на 100 г мелассы по формуле
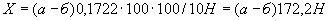 ,
,
где - содержание калия, %; - масса фильтра ТФ-4-20 с осадком после высушивания, г; - масса фильтра ТФ-4-20 без осадка, г;  - масса полученного осадка, г; - взятая навеска мелассы, г.
- масса полученного осадка, г; - взятая навеска мелассы, г.
Для пересчета на содержание KО надо найденное количество калия умножить на коэффициент 1,2046.
Пример. Масса фильтра с осадком =11,9635 г; масса фильтра без осадка =11,8725 г; взятая навеска мелассы =5,0425 г.
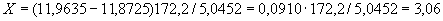 %.
%.
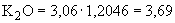 %.
%.
Для подготовки стеклянных фильтров для дальнейших анализов осадок смывают и кипятят фильтры в дистиллированной воде 30 мин. Затем воду сливают и в каждый фильтр наливают крепкой серной кислоты (фильтры устанавливают в химическом стакане). Спустя 1 ч кислоту сливают, фильтры промывают, кипятят в чистой воде 30 мин и промывают до полного удаления кислоты. Чистый фильтр высушивают при 105 °С до постоянной массы.
Определение суммы солей кальция и магния
Содержание кальциевых солей в мелассе колеблется от 0,2 до 2,5% в пересчете на СаО окиси магния MgO - от 0,001 до 1,2%. Навеску мелассы в количестве 1 г или определенный объем раствора мелассы помещают в коническую колбочку вместимостью 200-250 мл, в которую добавляют определенный объем дистиллированной воды (100 мл).
Пробу хорошо размешивают вращательным движением колбочки и к полученному раствору добавляют 5 мл аммиачного буферного раствора. Затем из бюретки добавляют избыток (10-15 мл) комплексообразователя трилона Б (н/28) и добавляют 7-8 капель индикатора хромогена-черного, после чего жидкость окрашивается в зеленый цвет. Эту жидкость титруют из другой бюретки магнезиальной смесью (н/28) до появления винно-красного цвета, который образуется вследствие полного связывания ионов кальция и магния (см. приложение II).
Для определения количества солей кальция и магния в дистиллированной воде, которая была добавлена, в коническую колбочку вместимостью 200-250 мл наливают 100 мл дистиллированной воды и таким же способом определяют содержание в ней солей кальция и магния.
Количество трилона Б, пошедшее на титрование 100 мл дистиллированной воды, вычитают из общего количества трилона Б, израсходованного при определении солей кальция и магния в навеске мелассы или в растворе.
Содержание солей кальция и магния вычисляют по формуле
 ,*
,*
где - общее количество трилона Б, мл; - количество трилона Б, пошедшее на титрование воды, мл; - поправочный коэффициент трилона Б;  и
и  - количество магнезиальной смеси, пошедшее на обратное титрование контроля и опыта, мл; - навеска испытуемого продукта, г; 1000 - коэффициент для пересчета миллиграммов в граммы.
- количество магнезиальной смеси, пошедшее на обратное титрование контроля и опыта, мл; - навеска испытуемого продукта, г; 1000 - коэффициент для пересчета миллиграммов в граммы.
________________
* Соответствует оригиналу. Примечание изготовителя базы данных.
Пример. 1 г мелассы растворяют в 100 мл дистиллированной воды. К полученному раствору мелассы из бюретки добавляют 10 мл трилона Б (н/28), поправочный коэффициент равен 0,99. На обратное титрование израсходовано магнезиальной смеси 8,5 мл (поправочный коэффициент равен 1). Трилона Б на связывание ионов кальция и магния пошло
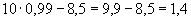 мл.
мл.
При определении содержания солей кальция и магния в 100 мл дистиллированной воды пошло 2 мл трилона Б (н/28), поправочный коэффициент равен 0,99. На обратное титрование магнезиальной смеси (н/28) пошло 1,8 мл (=1).
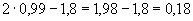 .
.
Содержание кальциевых и магниевых солей (в % к массе мелассы) определяют по формуле
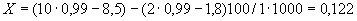 %.
%.
Для определения содержания солей кальция в мелассе применяют прямой метод определения содержания магния в мелассе и полученное его количество вычитают из суммы солей (Са+Mg), рассчитанной приведенным выше способом.
Определение содержания магния
Навеску мелассы 0,5 г переносят со 100 мл дистиллированной воды в колбочку вместимостью 250 мл, хорошо размешивают до полного растворения мелассы, нейтрализуют несколькими каплями соляной кислоты (1:1) до перехода бумажки конго к сине-фиолетовому цвету.
Для осаждения ионов кальция добавляют 5-10 мл насыщенного раствора щавелевокислого аммония, энергично взбалтывают 2-3 мин и оставляют на холоде в течение 15-20 мин. Затем добавляют 10 мл аммиачного буферного раствора, 10-15 мл комплексообразователя трилона Б (0,05 н.), сухого индикатора хромогена-черного (на кончике стеклянной палочки) и титруют 0,05 н. раствором сернокислого магния до перехода окраски индикатора от зеленого в оранжево-красный.
Содержание магния (в %) рассчитывают по формуле
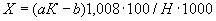 ,
,
где - количество MgO, %; - количество трилона Б, взятое на определение, мл; - поправочный коэффициент трилона Б; - количество 0,05 н. раствора сернокислого магния, пошедшее на титрование, мл; - навеска мелассы, г; 1000 - перевод миллиграммов в граммы; 1,008 - коэффициент перевода миллилитров трилона Б в MgO (1 мл 0,05 н. раствора трилона Б соответствует 1,008 мг MgO).
Определение буферности мелассы
Буферность - это свойство мелассы противостоять изменению реакции (рН) под влиянием кислоты или щелочи. Буферность зависит от состава несахаров и их количества в мелассе, главным образом от соотношения солей калия, натрия, кальция и магния, а также от количества аминного азота.
Ход определения заключается в следующем. На технических весах в стаканчике вместимостью 100 мл отвешивают 25 г мелассы, добавляют 25 мл дистиллированной воды и тщательно перемешивают до полного растворения ее. В растворе определяют рН. Затем из бюретки постепенно приливают 1 н. раствор серной кислоты и определяют количество его, необходимое для изменения рН мелассного раствора до 4,5 или 5. Израсходованное количество миллилитров 1 н. раствора серной кислоты умножают на 4, получают расход ее на 100 г мелассы.
Мелассы с нормальной буферностью характеризуются постепенным снижением рН мелассного раствора в пределах 7-5 при затрате на титрование по 0,8-0,6 мл 1 н. раствора серной кислоты, а до рН 4 - при дополнительном расходе 0,6-0,5 мл 1 н. раствора серной кислоты.
При повышенной буферности мелассы для снижения рН в пределах 7-5 требуется 1,2-2,3 мл 1 н. раствора серной кислоты, а для снижения рН до 4 расходуется 2-4 мл 1 н. раствора серной кислоты.
Буферность мелассы имеет значение при осветлении ее по холодно-кислотному способу и подкислении до рН 4,5-5. Для мелассы нормальной буферности расход серной кислоты меньше.
Определение расхода серной кислоты для осветления мелассы
На технических весах в стаканчике вместимостью 100 мл отвешивают 25 г мелассы, добавляют 25 мл дистиллированной воды, тщательно перемешивают до полного растворения мелассы и определяют рН раствора. Затем из бюретки приливают постепенно 1 н. раствор серной кислоты и определяют количество ее, необходимое для сдвига рН мелассного раствора до значения рН 4,5-5.
Расход серной кислоты на 1 т мелассы вычисляют исходя из найденного количества 1 н. раствора серной кислоты, пошедшего на титрование 25 г мелассы.
Пример. На титрование 25 г мелассы израсходовано 6,9 мл 1 н. раствора серной кислоты, что соответствует 6,9·0,049=0,338 г 100%-ной концентрированной серной кислоты, а на 1 кг мелассы расход серной кислоты составит 0,338·1000/25=13,5 г; на 1 т мелассы потребуется 13,5 кг 100%-ной серной кислоты или 14,4 кг (относительной плотностью 1,83).
Определение цветности мелассы
Цветность определяют путем сравнения окраски 2%-ного раствора мелассы с окраской раствора йода и выражают ее в миллилитрах 0,1 н. раствора йода. Обычно цветность нормальной мелассы соответствует 2 мл 0,1 н. раствора йода.
Отвешивают в химическом стакане 20 г мелассы и при помощи горячей дистиллированной воды смывают ее в мерную колбу вместимостью 100 мл. Раствор после охлаждения доводят до метки, 10 мл этого раствора разводят в мерной колбе вместимостью 100 мл в 10 раз, доливая содержимое колбы до метки водой, взбалтывают и фильтруют. 50 мл фильтрата помещают в стаканчик, в другой стаканчик такого же объема наливают 47 мл дистиллированной воды, стаканчик ставят против света, в стаканчик с водой наливают из бюретки 0,1 н. раствора йода до выравнивания окраски жидкости в обоих стаканчиках.
Количество миллилитров 0,1 н. раствора йода умножают на 2, чтобы выразить цветность мелассы по отношению к 100 мл 2%-ного раствора ее.
Определение содержания летучих кислот методом отгонки с водяным паром
К летучим кислотам мелассы относятся масляная, муравьиная, уксусная, валериановая, пропионовая. Эти кислоты в свободном состоянии даже в небольших количествах (0,006-0,008%) угнетают рост дрожжей. Большинство летучих кислот в мелассе находится в форме солей, которые менее вредны, чем свободные кислоты.
Содержание летучих кислот в свекловичной мелассе за последние пять лет колеблется в пределах от 0,33 до 1,15%. Оно не должно превышать 1,2%.
Количественное определение летучих кислот в мелассе основано на разложении солей этих кислот серной или фосфорной кислотой с последующей отгонкой их с водяным паром и определением их в дистилляте титрованием щелочи.
Для определения отвешивают 20 г мелассы и растворяют в 200 мл дистиллированной воды в колбе вместимостью 500 мл (рис.17), в которую прибавляют по каплям (до посинения бумажки конгорот) 0,5-1,0 мл серной кислоты (относительной плотностью 1,84) или 1,0 мл фосфорной кислоты (относительной плотностью 1,66) и закрывают ее резиновой пробкой. В пробке имеется два отверстия, через которые проходят стеклянные трубки: одна соединяется с парообразователем 1 и доходит почти до дна колбы 2 , а другая кончается у нижнего края пробки и соединяется с холодильником. Эта трубка служит для отвода дистиллята из колбы 2 в приемную колбу 3 вместимостью 250 мл. В качестве парообразователя применяют или специальный медный (или жестяной) сосуд, или стеклянную колбу вместимостью 2-3 л.
Парообразователь закрывают пробкой с длинной стеклянной трубкой, доходящей до дна сосуда; эта трубка служит предохранителем для предотвращения случайного повышения давления в процессе работы. Сначала воду в парообразователе 1 нагревают до кипения, затем его соединяют с перегонной колбой, содержимое которой предварительно подогревают. Летучие кислоты отгоняют при одновременном подогревании колбы и парообразователя. Нагревание парообразователя регулируют так, чтобы объем жидкости в отгонной колбе не увеличивался от конденсации поступающего пара и оставался постоянным. Отгоняют 5-6 порций дистиллята по 200 мл и титруют каждую партию 0,1 н. раствором гидроксида натрия в присутствии фенолфталеина.
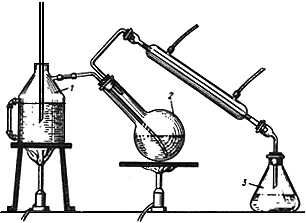
Рис.17. Прибор для определения летучих кислот
Отгонку летучих кислот заканчивают при получении двух следующих одна за другой порций дистиллята, имеющих одинаковую кислотность. Из количества миллилитров щелочи, пошедшего на титрование каждой порции отгона, вычитают количество миллилитров щелочи, израсходованное на титрование последней порции. Полученное таким образом количество миллилитров щелочи, затраченное на титрование каждой порции, кроме последней, складывают, умножают на 5 и на 0,006 и получают количество летучих кислот в 100 г мелассы в пересчете на уксусную кислоту.
Содержание летучих кислот в мелассе (в %) определяют по формуле
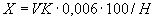 ,
,
где - количество 0,1 н. раствора гидроксида натрия, затраченное на титрование каждой порции, кроме последней, мл; 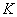 - поправочный коэффициент к 0,1 н. раствору гидроксида натрия; 0,006 - содержание уксусной кислоты, соответствующее 1 мл 0,1 н. раствора гидроксида натрия, г; 100 - пересчет на проценты; - навеска мелассы, г.
- поправочный коэффициент к 0,1 н. раствору гидроксида натрия; 0,006 - содержание уксусной кислоты, соответствующее 1 мл 0,1 н. раствора гидроксида натрия, г; 100 - пересчет на проценты; - навеска мелассы, г.
Пример. Количество 0,1 н. раствора гидроксида натрия, затраченное на титрование каждой порции, кроме последней, равно 31,1 мл, поправочный коэффициент к щелочи =1,0052, навеска мелассы 20 г.
Подставляя значения в формулу, определяют содержание летучих кислот в мелассе
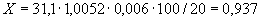 %.
%.
Ускоренный метод определения содержания летучих кислот (ХО ВНИИХП)
Прибор для отгонки летучих кислот (рис.18) состоит из широкогорлой конической колбы для отгонки 1 вместимостью 1,5-2 л, служащей парообразователем, холодильника 2 , приемной колбы 3 , перегонного аппарата типа эбулиостата, представляющего собой пробирку 4 . Общая длина пробирки 28 см, длина суженой части 6,5 см. В верхней части пробирки впаяна доходящая почти до дна тонкая изогнутая трубка 5 .
Рис.18. Прибор для определения летучих кислот ускоренным методом
Пробирка 4 соединена с парообразователем 1 с помощью резиновой пробки с двумя отверстиями. В одно отверстие вставляют предохранительную стеклянную трубку, на которую надета каучуковая трубка 6 с винтовым зажимом, а на второе - суженую часть пробирки, соединяющуюся с каплеуловителем 7 . Последний соединен с холодильником, он устраняет опасность попадания в холодильник отдельных капелек кислого раствора мелассы, уносимых при барботировании с паром. Парообразователь примерно на 2/3 наполняют дистиллированной водой, чтобы во время кипения вода не заливала отверстия трубки 5 , устанавливают на электроплитку или другой нагревательный прибор, кипятят воду в течение 10-15 мин, после чего в кипящую воду помещают аппарат с раствором мелассы.
Отвешивают 50 г мелассы и количественно переводят с теплой дистиллированной водой в мерную колбу на 100 мл. Раствор охлаждают до 20 °С, доводят до метки, тщательно перемешивают и подкисляют 10%-ным раствором серной кислоты до рН 3-3,2. При этом определяют количество серной кислоты, пошедшее на подкисление.
В химический стаканчик вносят пипеткой 10 мл приготовленного раствора мелассы, что соответствует 5 г мелассы, и добавляют по 0,5 мл 10%-ный раствор серной кислоты, каждый раз замеряя активную кислотность (рН).
Затем приступают к отгонке летучих кислот. В чистый аппарат для отгонки осторожно по стенке вносят пипеткой установленное количество 10%-ной серной кислоты, затем туда же добавляют каплю олеиновой кислоты и вводят пипеткой 10 мл раствора мелассы. Двумя-тремя миллилитрами дистиллированной воды смывают верхнюю часть трубки аппарата, по которой вносили растворы.
Подготовленный таким образом отгонный аппарат помещают в колбу-парообразователь с кипящей водой, быстро соединяют с каплеуловителем и холодильником и только после этого закрывают зажим на предохранительной трубке 6 . Образующийся при кипении воды пар из колбы 1 через трубку 5 попадает в пробирку 4 и, барботируя раствор мелассы, уносит с собой летучие кислоты в холодильник.
Отгонка летучих кислот ведется в мерную колбочку на 50 мл. Длительность отгонки 50 мл составляет 10-15 мин.
По окончании отгонки отставляют приемную колбочку и открывают зажим на предохранительной трубке 6 . Содержимое мерной колбочки переводят в коническую колбочку, мерную колбочку дважды ополаскивают 5 мл свежекипяченой дистиллированной водой, сливают в коническую колбочку с отгоном и титруют 0,1 н. раствором гидроксида натрия в присутствии индикатора фенолфталеина до слабо-розового окрашивания.
При отсутствии мерных колбочек отгон можно собирать в обыкновенные конические колбочки на 100-150 мл, на которые предварительно наносят метку, соответствующую 50 мл. Титрование щелочью ведут в этих же колбочках.
Содержание летучих кислот рассчитывают на 100 г мелассы (сухое вещество) с условным пересчетом на уксусную кислоту.
Содержание летучих кислот (в %) определяют по формуле
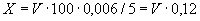 ,
,
где - количество 0,1 н. раствора гидроксида натрия, пошедшее на титрование 50 мл отгона, мл; 0,006 - количество уксусной кислоты, соответствующее 1 мл 0,1 н. раствора гидроксида натрия; 5 - навеска мелассы, г.
Пример. На титрование 50 мл отгона пошло 6,8 мл 0,1 н. раствора гидроксида натрия. Следовательно, в 100 г мелассы содержится летучих кислот 6,3·0,12=0,816%.
Для пересчета на сухое вещество мелассы нужно найденное количество летучих кислот разделить на содержание сухих веществ и умножить на 100.
В мелассе содержится 74,5% СВ. Тогда количество летучих кислот, считая на сухое вещество, составит 0,816·100/74,5=1,1%.
Определение содержания сернистого ангидрида
Содержание сернистого ангидрида в мелассе не должно превышать 0,05%. Наличие сернистого ангидрида в мелассе тормозит рост дрожжей, а прессованные дрожжи, выращенные на такой мелассе, имеют свойство быстро изменять свое качество и темнеть при хранении.
Сущность метода определения состоит в окислении йодом сернистой кислоты в серную.
Колбу (рис.19) 1 для отгона вместимостью 500 мл закрывают каучуковой пробкой с двумя отверстиями. В одно отверстие вставляют воронку 4 для ввода исследуемой жидкости, другое отверстие сообщается посредством каплеуловителя с одним концом холодильника 3 , другой конец которого должен быть опущен в приемную колбу 2 .
Рис.19. Прибор для определения сернистого ангидрида
В колбу для отгона наливают 20 г мелассы, разбавляют 250-300 мл воды и добавляют 15 мл 25%-ного раствора ортофосфорной кислоты. В приемную колбу отмеривают точно 25 мл 0,1 н. раствора йода и помещают в баню со льдом.
Конец холодильника погружают в йодный раствор, дистилляцию ведут на медленном огне. Отгонку продолжают до тех пор, пока половина жидкости будет отогнана до прекращения выделения пузырьков газа. Общая длительность отгонки должна быть не менее 2,5 ч. Затем холодильник смывают дистиллированной водой в приемную колбу и количество йодного раствора, не вошедшее в реакцию, титруют 0,1 н. раствором гипосульфита в присутствии нескольких капель 1%-ного раствора крахмала.
Одновременно для установления поправки на окисляемость йодом находящихся в растворах веществ проводят глухой опыт с таким же количеством реактивов, но без навески мелассы. Продолжительность глухого опыта должна соответствовать продолжительности отгонки.
Содержание сернистого ангидрида (в %) в мелассе определяют по формуле
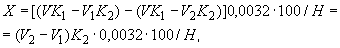 ,
,
где - количество 0,1 н. раствора йода, мл; 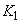 - поправочный коэффициент к 0,1 н. раствору йода; - количество 0,1 н. раствора гипосульфита, израсходованное на титрование, мл; - количество 0,1 н. раствора гипосульфита, израсходованное в глухом опыте, мл;
- поправочный коэффициент к 0,1 н. раствору йода; - количество 0,1 н. раствора гипосульфита, израсходованное на титрование, мл; - количество 0,1 н. раствора гипосульфита, израсходованное в глухом опыте, мл; 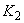 - поправочный коэффициент к 0,1 н. раствору гипосульфита; 0,0032 - содержание SO, соответствующее 1 мл 0,1 н. раствора йода, г; 100 - пересчет в проценты; - навеска мелассы, г.
- поправочный коэффициент к 0,1 н. раствору гипосульфита; 0,0032 - содержание SO, соответствующее 1 мл 0,1 н. раствора йода, г; 100 - пересчет в проценты; - навеска мелассы, г.
На анализ всегда берут 20 г мелассы и 25 мл 0,1 н.раствора йода. Подставляя эту величину в уравнение, получим его в упрощенном виде
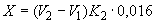 .
.
Пример. Для определения SO взято 20 г мелассы, в приемную колбу добавлено 25 мл 0,1 н. раствора йода. При титровании израсходовано 22,4 мл 0,1 н. раствора гипосульфита с поправочным коэффициентом =1,0197. Поправочный коэффициент 0,1 н. раствора йода =1. На глухой опыт израсходовано 24,1 мл гипосульфита.
Подставляя значения в формулу, определяют содержание сернистого ангидрида в мелассе
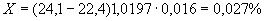 .
.
Определение содержания коллоидов
Повышенное содержание коллоидов в мелассе вызывает сильное вспенивание в процессе переработки. Допустимым является содержание коллоидов, равное 4-7% к мелассе.
Содержание коллоидов в мелассе определяют по методу А.В.Думанского и С.Е.Харина. 15 г мелассы разбавляют дистиллированной водой до содержания сухих веществ, равного 10%. В случае щелочной реакции раствор нейтрализуют соляной кислотой по фенолфталеину. На 100 мл раствора мелассы добавляют 5 мл 0,1 н. раствора соляной кислоты, при этом колебание рН полученного раствора допускается в пределах 4-6.
Подкисленный раствор мелассы разбавляют дистиллированной водой до содержания сухих веществ, равного 5%, и из вновь разбавленного раствора отбирают на определение коллоидов 5 мл в колбочку вместимостью 100 мл, куда заранее наливают смесь, состоящую из 50 мл этилового спирта и 2,5 мл серного эфира.
Колбочку закрывают корковой пробкой, через которую проходит обратный холодильник, погружают в водяную баню, нагретую до кипения. Кипение содержимого колбы поддерживают в течение 3-4 мин. После охлаждения содержимое колбы фильтруют через заранее высушенный до постоянной массы плотный беззольный или фильтрующий тигель ТФ-4-20.
Осадок на фильтре промывают 80-100 мл смеси, состоящей из 100 мл спирта, 5 мл эфира и 10 мл дистиллированной воды. Промытый осадок вместе с фильтром переносят в бюкс и высушивают до постоянной массы при температуре 100-101 °С. Количество коллоидов выражают в процентах к массе.
Пример. 15 г мелассы, взятые для определения коллоидов, разбавлены дистиллированной водой до содержания сухих веществ, равного 10%. Получено 110 мл раствора, к которому добавлено 5,5 мл 0,1 н. раствора НСl. Общий объем равен 115,5 мл, 50 мл полученного раствора разбавлено водой вдвое (до 100 мл), на определение взято 5 мл этого раствора.
Таким образом, в реакции осаждения коллоидов участвовало мелассы
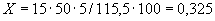 г.
г.
Количество высушенного осадка коллоидов равно 0,0205 г. В 0,325 г мелассы содержится 0,0215 г коллоидов; в 100 г равно 6,3 г (0,0205·100/0,325) или в исследуемой мелассе содержится 6,3% коллоидов.
Определение пенистости мелассы
20 мл раствора мелассы, содержащего 1,5% СВ, подкисляют до рН 4,7-5, помещают в мерный цилиндр с притертой пробкой вместимостью 50 мл и энергично встряхивают в течение 1 мин. Через 30 с после окончания встряхивания определяют высоту столба пены. Пенообразовательная способность мелассы характеризуется высотой столба пены, которая у нормальной мелассы должна быть не более 40 мм. Образовавшаяся пена постепенно исчезает. Время, в течение которого пена оседает на 10 мм, определяет стабильность ее. Продолжительность оседания пены должна быть не более 10 мин.
Определение содержания нефтяных продуктов
Метод основан на экстрагировании нефтяных продуктов, содержащихся в мелассе, петролейным эфиром с последующим отделением эфира и высушиванием полученного остатка до постоянной массы. Если в мелассе содержатся одновременно жиры, то их необходимо предварительно удалить омылением 0,1 н. раствором гидроксида натрия.
В коническую колбу вместимостью 150-200 мл отвешивают 50 г неразбавленной мелассы, добавляют 15-20 мл 0,1 н. KОН до ясной щелочной реакции и производят омыление, нагревая колбу в течение 30 мин на кипящей водяной бане. Полученный раствор переносят в делительную воронку вместимостью 300 мл, приливают 100 мл петролейного эфира (фракция петролейного эфира, перегоняемая при температуре до 60 °С) и взбалтывают в течение 30-40 мин до образования однородной смеси.
Смесь для расслаивания оставляют на несколько часов. При задержке расслаивания рекомендуется добавить 1-2 мл этилового спирта. Нижний слой, который представляет собой разбавленную мелассу, удаляют. Эфирный слой сливают в предварительно взвешенную колбу или бюкс. Удалив эфир на водяной бане, остаток высушивают при 60 °С до постоянной массы и взвешивают. Путем умножения массы на 2 получают количество нефтяных продуктов в мелассе в процентах.
Ускоренный метод оценки степени пригодности мелассы для дрожжевого производства по скорости роста дрожжей (технологическая микропроба)
При выращивании дрожжей очень важно, чтобы скорость накопления биомассы была устойчивой и стабильной при переработке мелассы разного состава. Поэтому при оценке степени пригодности мелассы для дрожжевого производства необходимо выявлять показатель скорости роста дрожжей на исследуемой мелассе. Однако в условиях заводских лабораторий химическими анализами невозможно быстро установить содержание в мелассе всех компонентов, стимулирующих или подавляющих рост дрожжей. Поэтому во ВНИИХПе разработан ускоренный метод определения степени пригодности мелассы для дрожжевого производства по скорости роста дрожжей (технологическая микропроба).
Наиболее точные результаты по определению скорости роста дрожжей в зависимости от состава мелассы получаются при выращивании дрожжей в течение первых 6 ч. В этот период нет недостатка в питательных веществах и кислороде для нормального размножения дрожжей.
Сущность метода заключается в культивировании 30 г дрожжей в течение 6 ч в лабораторном дрожжерастильном аппарате с расходом 90 г мелассы, содержащей 46% сахара и добавлением азот- и фосфорсодержащих солей (6,65 г сернокислого аммония и 2,64 г диаммонийфосфата).
После шестичасового выращивания определяют количество полученных дрожжей на исследуемой мелассе и скорость роста дрожжей 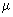 - натуральный логарифм отношения полученных дрожжей к засевным в единицу времени, который вычисляют по формуле
- натуральный логарифм отношения полученных дрожжей к засевным в единицу времени, который вычисляют по формуле
 ,
,
где - скорость роста дрожжей; - биомасса, накопленная за определенный промежуток времени, г; - биомасса, взятая для засева, г; - продолжительность выращивания дрожжей.
Для выращивания дрожжей используется дрожжерастильный аппарат 1 (рис.20) из нержавеющей стали полезной вместимостью 3 л с воздухораспределительной системой 2 . Для поддержания необходимой температуры во время размножения дрожжей аппарат помещают в водяную баню 3 с контактным термометром 4 , установленную на электроподогрев 5 . Питательные вещества (меласса, растворы сернокислого аммония, диаммоний фосфата и аммиачная вода) поступают из сосудов 6 , 7 , 8 , 9 , установленных над аппаратом. Приток питательной среды регулируют при помощи программного регулирующего устройства или кранов по указателю уровня в соответствии с графиком расхода мелассы и питательных солей. Для аэрации среды в дрожжерастильный аппарат по воздуховоду 10 подают воздух, расход которого учитывают по счетчику 11 .
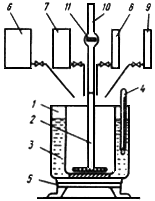
Рис.20. Установка для выращивания дрожжей по воздушно-приточному методу
Подготовка среды. Осветление мелассы производят по холодно-кислотному способу. Для этой цели отвешивают и помещают в стакан навеску мелассы (90 г), содержащей 46% сахара. К навеске добавляют 180 мл воды, содержащей серную кислоту (относительной плотностью 1,84), из расчета 0,6 мл кислоты на 100 г мелассы. Содержимое перемешивают 30 мин. Затем доводят объем осветленной мелассы до 360 мл и оставляют стоять 10-15 ч, по истечении которых мелассу декантируют и осадок отфильтровывают.
Расчет количества фосфора и азота для питания дрожжей. Питательные соли (сернокислый аммоний и диаммонийфосфат) расходуют в том же количестве, как и при выращивании дрожжей, по 10-часовой схеме, поскольку все питательные соли подаются в первый период выращивания, т.е. за 6 ч. В этот период растущие дрожжи не должны испытывать недостатка в азотном и фосфорном питании, в противном случае это может отразиться на скорости роста.
Расчет фосфорного питания проводят из расчета содержания в полученных дрожжах после опыта 1,1% PO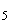 , ожидаемый выход на сырье принимается равным 80%, содержание РО в диаммонийфосфате 50%.
, ожидаемый выход на сырье принимается равным 80%, содержание РО в диаммонийфосфате 50%.
Для ожидаемого получения по 10-часовой схеме 120 г дрожжей из 150 г мелассы необходимо иметь в питательной среде 1,32 г РО. Это количество фосфора содержится в 2,64 г диаммонийфосфата. Навеску диаммонийфосфата (2,64 г) растворяют в 100 мл воды.
Расчет азотного питания проводят из расчета содержания в полученных дрожжах 1,8% азота. Предварительно определяют количество аминного азота в исследуемой мелассе. Для данного примера содержание аминного азота в мелассе принимается равным 0,2%.
Для обеспечения 120 г дрожжей из 150 г мелассы азотным питанием необходимо иметь в питательной среде 2,16 г азота. В 150 г мелассы содержится 0,3 г аминного азота, в 2,16 г диаммонийфосфата содержится 0,53 г азота (2,64:5), следовательно, недостает 1,33 г азота (2,16-0,8). Это количество азота добавляют в виде сернокислого аммония, содержащего 20% азота. При этом требуется добавить 6,65 г.
В дрожжерастильный аппарат наливают 1000 мл воды, подогретой до 30-35 °С, в соответствии с графиком притока мелассы добавляют в нее 30 мл осветленной мелассы, 30 г дрожжей, предварительно активированных при температуре 30 °С в течение 20 мин в смеси воды и осветленной мелассы (50+30 мл). Затем подают воздух для аэрации среды и наливают растворы диаммонийфосфата и сернокислого аммония. Через час подают осветленный мелассный раствор и питательные соли (растворы сернокислого аммония и диаммонийфосфата) по графику. Воду добавляют с таким расчетом, чтобы к концу опыта объем культуральной среды составил 2,5-2,7 л. Схема притока мелассы солей и воды приведена в табл.10.
Таблица 10
|
Компоненты |
Время притока, ч |
||||||
|
0-1 |
1-2 |
2-3 |
3-4 |
4-5 |
5-6 |
всего |
|
|
Меласса, % |
17 |
10 |
13 |
17 |
20 |
23 |
100 |
|
Меласса, г |
15 |
9 |
12 |
15 |
18 |
21 |
90 |
|
Осветленный мелассный раствор, мл |
60 |
36 |
48 |
60 |
72 |
84 |
360 |
|
Питательные соли, % |
20 |
20 |
20 |
20 |
20 |
20 |
100 |
|
Вода, мл |
1000 |
500 |
500 |
500 |
500 |
500 |
3500 |
Температуру среды поддерживают в пределах 30-32 °С, рН культуральной среды - 4,3-5,5, в случае снижения активной кислотности до 4 вместо сернокислого аммония добавляют аммиачную воду. Пену гасят олеиновой кислотой. В процессе выращивания дрожжей (за 6 ч) масса дрожжей в зависимости от качества мелассы увеличивается от 1,5 до 3 раз.
Для определения скорости роста дрожжей на исследуемой мелассе через 6 ч от начала выращивания определяют объем среды, устанавливают количество дрожжей с содержанием 25% СВ в 1 л среды и подсчитывают общее количество дрожжей, выросших в аппарате за 6 ч.
Скорость роста дрожжей определяют по формуле, приведенной на с.81.
Ниже приведены примеры определения скорости роста дрожжей на неполноценной мелассе (опыт 1) и на мелассе нормального состава (опыт 2).
Находим скорость роста дрожжей на исследуемой мелассе.
Опыт 1
|
|
|
|
|
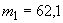 г
г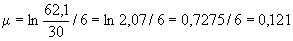
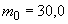 г
г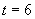 ч
ч
Опыт 2
|
|
|
|
|
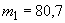 г
г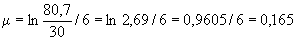
Ниже приведены данные о степени пригодности мелассы для дрожжевого производства по скорости роста дрожжей.
|
Качество мелассы |
Скорость роста дрожжей |
|
Нормальное |
0,148-0,170 |
|
Среднее |
0,124-0,135 |
|
Низкое |
0,105-0,115 |
Причины возникновения сахароаминной реакции и меры предупреждения ее в процессе приемки и хранения мелассы
Большое значение для дрожжевого производства имеет сохранение качества мелассы в процессе приемки, хранения и передачи по коммуникациям в подготовительное отделение завода.
Иногда причиной ухудшения качества мелассы может быть так называемая сахароаминная реакция, которая протекает в результате взаимодействия сахара с аминогруппами белков и аминокислот.
Известно, что при порче свеклы количество инвертного сахара может возрасти в 10 раз в результате инверсии сахарозы. Такое количество инвертного сахара благоприятно для возникновения сахароаминной реакции.
Внешние условия также имеют исключительно большое значение для возникновения и развития сахароаминной реакции. Так, следует отметить, что повышение температуры до 50-60 °С резко увеличивает скорость течения сахароаминной реакции. Такое повышение температуры может возникнуть при подогреве мелассы паром при перекачке зимой.
Протекание сахароаминной реакции в мелассе при хранении характеризуется подкислением (рН 6,2-5), увеличением инвертного сахара (4-10,3%) и снижением сахара по прямой поляризации (42,3-31,7%), иногда увеличением цветности до 5-8 мл 0,1 н. раствора йода против первоначальных показателей свежей мелассы. Следует отметить также, что все эти изменения протекают при небольшом содержании микроорганизмов (900-6000 в 1 г).
Результатом протекания сахароаминной реакции являются потеря сахара и ухудшение качества мелассы.
Для предупреждения сахароаминной реакции необходимо проводить следующие мероприятия:
-
для производства хлебопекарных дрожжей отбирать мелассу, полученную из здоровой свежей свеклы, т.е. в 1-м периоде сезона сахароварения (ноябрь, декабрь);
-
во избежание потерь сахара в результате взаимодействия его с аминокислотами мелассу хранить при возможно низкой температуре;
-
не допускать слива в хранилище мелассы, нагретой до температуры выше 40 °С;
-
обращать особое внимание на недопустимость произвольного нагрева мелассы в мелассохранилищах из-за неисправности запорной паровой коммуникации;
-
с целью постоянного контроля за температурой хранящейся мелассы в мелассохранилищах устанавливать термометры.
МЕТОДЫ АНАЛИЗА ОСНОВНЫХ МАТЕРИАЛОВ
Сульфат аммония (сернокислый аммоний)
В дрожжевом производстве в качестве основного источника азота используют сернокислый аммоний, или сульфат аммония (ГОСТ 10873-73). Ниже приведены физико-химические показатели сульфата аммония (NH)SO.
|
Показатели |
Норма |
|
Внешний вид |
Белые или слабо-желтые кристаллы |
|
Массовая доля основного вещества в сухом продукте, %, не менее |
|
|
в пересчете на сульфат аммония |
99 |
|
в пересчете на азот |
21 |
|
Массовая доля влаги, %, не более |
0,6 |
|
Массовая доля мышьяка, %, не более |
0,0005 |
|
Сульфиды и сульфиты, мл, не более |
0,5 (по высоте черного кольца) |
Очищенный сульфат аммония поступает на дрожжевые предприятия в недостаточном количестве, в связи с этим в дрожжевом производстве иногда применяют технический сульфат аммония. Он содержит повышенное количество вредных примесей (сульфидов и сульфитов). С целью определения пригодности сульфата аммония, поступающего на дрожжевые заводы, предлагается ускоренный метод определения сульфидов и сульфитов.
Методика определения сульфидов и сульфитов (солей сероводородной и сернистой кислот) ускоренным способом. В сосуд (рис.21) помещают 5 г сульфата аммония, наливают 50 мл дистиллированной воды, добавляют 3-4 капли 10%-ного раствора двуххлористого олова и 4 мл концентрированной, химически чистой или чистой для анализа серной кислоты.
Рис.21. Прибор для определения сульфидов и сульфитов
Насадку прибора заряжают следующим образом. В нижний конец трубочки насадки помещают кусочек чистой ваты, на которую накладывают вату, смоченную уксуснокислым свинцом. Все плотно утрамбовывают стеклянной палочкой. В верхний конец трубочки помещают чистый кусочек ваты. После зарядки трубочки в сосуд помещают 4 г металлического цинка. Сосуд немедленно закрывают заряженной насадкой и прибор ставят в темное место. За реакцией ведут наблюдение в течение 4 ч, после чего измеряют величину темного кольца на ватке, смоченной уксуснокислым свинцом, возникающего при наличии вредных примесей в сульфате аммония.
Если величина темного кольца на ватке, смоченной уксуснокислым свинцом, превышает 0,5 мм, т.е. сульфат аммония содержит вредные примеси, перед его использованием производят выдерживание на воздухе.
В процессе приготовления раствора из такого сульфата аммония проводят его выстаивание в течение 4 ч или продувание воздухом в течение 2 ч.
Определение массовой доли влаги. Около 5 г сульфата аммония взвешивают с точностью до 0,0002 г и сушат в сушильном шкафу при 100-110 °С до постоянной массы. Массовую долю влаги выражают в процентах.
Определение массовой доли сульфата аммония. 20 г сульфата аммония растворяют в воде (дистиллированной), переносят в мерную колбу вместимостью 500 мл и доводят водой до метки. Из полученного раствора отбирают пипеткой 25 мл и нейтрализуют 0,1 н. раствором гидроксида натрия с индикатором метиловым красным. Затем прибавляют 25 мл формалина, предварительно нейтрализованного 33%-ным раствором гидроксида натрия в присутствии фенолфталеина. Свободную серную кислоту, выделившуюся при этом, титруют 0,5 н. раствором гидроксида натрия в присутствии фенолфталеина.
Массовую долю сульфата аммония (в %) в пересчете на сухое вещество определяют по формуле
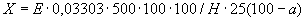 ,
,
где - объем точно 0,5 н. раствора гидроксида натрия, пошедший на титрование сульфата аммония, мл; - навеска сернокислого аммония, г; - массовая доля влаги в пробе, %; 0,03303 - количество сульфата аммония, соответствующее точно 1 мл 0,5 н. раствора гидроксида натрия, г.
Для пересчета на азот процентное содержание сульфата аммония умножают на 0,212.
Определение массовой доли мышьяка. 5 г сульфата аммония взвешивают с точностью до 0,001 г и ведут определение по методике, описанной на с.96.
Аммиак водный
Аммиак применяют одновременно как источник азота и для регулирования рН среды при выращивании дрожжей. В дрожжевом производстве используют аммиак водный технический (ГОСТ 9-77*) - раствор синтетического аммиака в воде. Водный аммиак выпускают марок А и Б. Для дрожжевого производства используют аммиак марки А. Определяется содержание аммиака.
______________
* На территории Российской Федерации действует ГОСТ 9-92. - Примечание изготовителя базы данных.
Процентное содержание аммиака в водном растворе определяют по относительной плотности и титрованием. Относительную плотность аммиачного раствора определяют пикнометром или ареометром, затем устанавливают процентное содержание аммиака.
При определении содержания аммиака титрованием отбирают при помощи бюретки в мерную колбу вместимостью 200 мл 10 мл испытуемого раствора, доливают до метки дистиллированной водой и раствор тщательно перемешивают. 5 мл полученного раствора отбирают пипеткой и переносят в колбу для титрования, доводят дистиллированной водой до 50 мл и титруют 0,1 н. раствором серной кислоты с индикатором метиловым красным до розового оттенка. Количество миллилитров кислоты, затраченное на титрование и умноженное на 0,0017 (количество граммов аммиака, соответствующее 1 мл 0,1 н. раствора серной кислоты), дает содержание аммиака в 5 мл раствора. Содержание аммиака в испытуемом растворе определяют по формуле
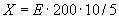 ,
,
где - количество аммиака в 5 мл раствора, взятое для титрования, г.
Диаммонийфосфат
Диаммонийфосфат (NH)НРО используют как источник фосфора и азота. Диаммонийфосфат технический изготовляют двух марок (А и Б) со следующими показателями.
|
Показатели |
Диаммонийфосфат марки |
||
|
А |
Б |
||
|
Внешний вид и цвет |
Кристаллы белого цвета |
Кристаллы белого цвета, допускается сероватый или желтоватый оттенок |
|
|
Фосфорный ангидрид в пересчете на Р |
52,0 |
51,0 |
50,0 |
|
Аммиак, %, не менее |
23,5 |
23,0 |
22,5 |
|
Влага, %, не более |
4,0 |
5,0 |
5,0 |
|
Мышьяк, %, не более |
0,002 |
Не нормируется |
|
|
Фтор, %, не более |
0,01 |
0,05 |
Не нормируется |
Определяется содержание фосфорного ангидрида, аммиака, мышьяка, влаги, фтора.
Определение содержания массовой доли фосфорного ангидрида. Отвешивают в стаканчик 1,0-1,2 г диаммонийфосфата с погрешностью не более 0,001 г и количественно переносят в мерную колбу вместимостью 500 мл. При 20 °С доводят водой до метки, смесь перемешивают и 10 мл отбирают в мерную колбочку на 100 мл. Доводят водой до метки, перемешивают, на определение фосфата отбирают в пробирку 1 мл и ведут определение калориметрическим методом Бригса.
Определение массовой доли аммиака. Навеску около 2,5 г продукта взвешивают с точностью до 0,001 г, переводят в мерную колбу вместимостью 250 мл, растворяют дистиллированной водой и объем доводят до метки. Из раствора отбирают пипеткой 25 мл и переносят в коническую колбу вместимостью 150 мл, сюда же прибавляют 2-3 капли фенолфталеина и нейтрализуют 0,1 н. раствором гидроксида калия до слабо-розового окрашивания. После этого добавляют 10-15 мл нейтрализованного раствора формалина (40%-ного) и вторично титруют 0,1 н. раствором гидроксида калия до появления слабо-розового окрашивания.
Количество аммиака (в %) определяют по формуле
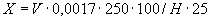 ,
,
где - объем 0,1 н. раствора гидроксида калия, пошедший на титрование, мл; 0,0017 - количество NH, соответствующее 1 мл 0,1 н. раствора гидроксида калия, г; - навеска испытуемого продукта - диаммонийфосфата, г.
Определение массовой доли влаги.
5 г диаммонийфосфата отвешивают на аналитических весах и сушат в термостате при температуре 60 °С до постоянной массы. Содержание массовой доли 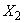 (в %) определяют по формуле
(в %) определяют по формуле
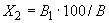 ,
,
где  - содержание массовой доли влаги в навеске после высушивания, г;
- содержание массовой доли влаги в навеске после высушивания, г;  - масса навески до высушивания, г.
- масса навески до высушивания, г.
Определение массовой доли мышьяка. 5 г продукта отвешивают с точностью до 0,001 г и ведут определение по методике (см. с.96).
Определение массовой доли фтора. Содержание водорастворимого фтора в диаммонийфосфате допускается в количестве не выше 0,01%.
Наличие водорастворимого фтора угнетающе действует на процесс размножения дрожжей, поэтому контроль за соблюдением указанных норм имеет большое значение. Количество фтора в среде дрожжерастильного аппарата не должно превышать 0,001%.
Метод определения фтора основан на приводимых ниже реакциях, происходящих при смешивании исследуемого вещества с кварцевым песком, с разбавленной серной кислотой и при нагревании.
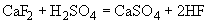 ;
;
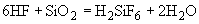 ;
;
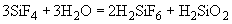 .
.
При избыточном количестве SiO кремнефтористоводородная кислота подвергается дистилляции, количественно переходя в приемник; чистая HSiF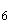 улетучивается при 40 °С без остатка, причем отношение SiF:HF меняется в зависимости от концентрации HSiF. Обычно остаток обогащается HF, но при наличии SiO превращается в новое количество HSiF и т.д. Дистиллят смешивают с KCl, причем выделяется KSiF, который может быть отфильтрован; осадок растворяют горячей водой и раствор титруют щелочью.
улетучивается при 40 °С без остатка, причем отношение SiF:HF меняется в зависимости от концентрации HSiF. Обычно остаток обогащается HF, но при наличии SiO превращается в новое количество HSiF и т.д. Дистиллят смешивают с KCl, причем выделяется KSiF, который может быть отфильтрован; осадок растворяют горячей водой и раствор титруют щелочью.
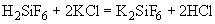 ;
;
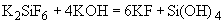 .
.
Для определения берут навеску около 1,5 г исследуемого материала и 2 г прокаленного морского песка; все это помещают в круглодонную колбу 1 для отгонки (рис.22), соединенную при помощи трубки 2 с шариковым холодильником, конец которого опущен в коническую приемную колбу 3 вместимостью 100-150 мл. На дно приемной колбочки помещают 3-4 г химически чистого хлористого калия, предварительно измельченного (растертого) в ступке. В круглодонную колбу наливают 30 мл серной кислоты, разбавленной водой 1:3, и колбу подогревают. Приемную колбу, в которую поступает отгон, время от времени встряхивают.
Рис.22. Прибор для определения фтора
Отгонку ведут до тех пор, пока в круглодонной колбе не останется 5-10 мл жидкости и не появятся белые пары SO, а отгона наберется 20-25 мл. По окончании отгонки трубки и холодильник смывают 20-30 мл 96%-ного этилового спирта (в зависимости от количества отгона) до образования 50%-ного раствора спирта.
Приемную колбу закрывают пробкой и оставляют на 2-4 ч. Затем жидкость с осадком фильтруют через бумажный фильтр. Осадок промывают на фильтре 3-4 раза 20%-ным раствором хлористого калия, а затем 50%-ным раствором спирта до нейтральной реакции по метиловому оранжевому (в фильтрате).
После промывания фильтр вместе с осадком опускают в чистую сухую коническую колбочку вместимостью 100-160 мл и растворяют горячей дистиллированной водой (без СО). Раствор осторожно доводят до кипения и тотчас же титруют 0,1 н. раствором гидроксида калия. В конце титрования кипячение повторяют, а раствор вновь титруют до розового окрашивания его с индикатором фенолфталеином.
Количество кремнефтористоводородной кислоты (в %) определяют по формуле
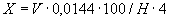 ,
,
где - количество 0,1 н. раствора щелочи, мл; - навеска вещества, г; 0,0144 - количество кремнефтористоводородной кислоты, соответствующее 1 мл 0,1 н. раствора щелочи (14,4/4 г выражает эквивалент 0,1 н. раствора кремнефтористоводородной кислоты, следовательно, 1 мл данного раствора соответствует 14,4/4·1000=0,0144/4 г кремнефтористоводородной кислоты).
Количество кремнефтористоводородной кислоты (в %) пересчитывают на количество содержащегося в ней фтора по формуле
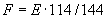 ,
,
где - количество кремнефтористоводородной кислоты, %; 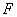 - количество фтора, %; 114 - молекулярная масса ; 144 - молекулярная масса кремнефтористоводородной кислоты НSiF.
- количество фтора, %; 114 - молекулярная масса ; 144 - молекулярная масса кремнефтористоводородной кислоты НSiF.
Кукурузный экстракт
Кукурузный экстракт используют в дрожжевом производстве как источник активаторов роста, в основном биотина. Кукурузный экстракт получают путем уваривания под вакуумом экстрактных замочных вод, являющихся побочным продуктом (отходом) кукурузнокрахмального производства.
Состав кукурузного экстракта характеризуется следующими показателями. По внешнему виду - густая непрозрачная жидкость с хлопьевидной взвесью, способной расслаиваться, или почти однородная паста. Цвет кукурузного экстракта от желтого до темно-коричневого, запах, характерный для кукурузы, содержание сухих веществ не менее 48%, кислотность (в % молочной кислоты) не менее 17,0%, свободного сернистого газа не более 0,35%, содержание золы не более 21,0% (в пересчете на СВ). В кукурузном экстракте определяются содержание сухих веществ и кислотность.
Арбитражный метод определения сухих веществ. Для определения сухих веществ данным методом бюксу с бумажными роликами высушивают до постоянной массы. Для изготовления бумажных роликов разрезают лист фильтровальной бумаги на полоски шириной 10 мм и длиной 500 мм и скатывают в нетугие ролики.
В бюксу кладут 3-4 ролика и высушивают до постоянной массы при температуре 105 °С. Сначала его помещают в сушильный шкаф на 3 ч, охлаждают в эксикаторе, взвешивают, а затем сушат по 30 мин до тех пор, пока разница между двумя последующими взвешиваниями не будет превышать 0,001 г.
Высушенные ролики опрокидыванием помещают в крышку бюксы. В бюксу без крышки берут навеску экстракта 1-1,5 г с погрешностью не более 0,0002 г. К навеске приливают примерно двойное количество дистиллированной воды, подогретой до 30-40 °С, и содержимое бюксы, помешивая круговым движением для равномерного растворения продукта. Затем в бюксе помещают ролики, в которые раствор должен полностью впитаться.
Бюксу с открытой крышкой ставят в сушильный шкаф и сушат в течение 4 ч при температуре 60 °С. После этого температуру в шкафу увеличивают до 105 °С и экстракт выдерживают при этой температуре сначала 6-8 ч, охлаждают в эксикаторе 30 мин, взвешивают, а затем сушат по 30 мин до тех пор, пока разница между двумя последующими взвешиваниями не будет превышать 0,001 г.
Содержание сухих веществ кукурузного экстракта (в %) рассчитывают по формуле
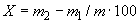 ,
,
где  - масса бюксы с роликами, г; - масса кукурузного экстракта до высушивания, г;
- масса бюксы с роликами, г; - масса кукурузного экстракта до высушивания, г;  - масса бюксы с экстрактом и роликами после высушивания, г.
- масса бюксы с экстрактом и роликами после высушивания, г.
Определение кислотности. Взвешивают 2 г экстракта с точностью 0,001 г, переносят количественно в мерную колбу вместимостью 200 мл, доводят нейтральной (предварительно оттитрованной перед определением кислотности до такой же окраски, как указано ниже) дистиллированной водой до метки при температуре 20 °С и хорошо перемешивают. 50 мл полученного раствора титруют 0,1 н. раствором гидроксида натрия до слабо-розового окрашивания в присутствии фенолфталеина до появления розовой окраски, не исчезающей в течение 1 мин.
Кислотность экстракта выражают в процентах молочной кислоты в пересчете на СВ экстракта и вычисляют по формуле
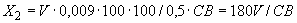 ,
,
где - количество 0,1 н. раствора гидроксида натрия, пошедшее на титрование, мл; - содержание сухих веществ в сгущенном кукурузном экстракте, %; 0,009 - количество молочной кислоты, соответствующее 1 мл 0,1 н. раствора гидроксида натрия, г; 0,5 - масса кукурузного сгущенного экстракта в объеме раствора, взятом на титрование, г.
Определение содержания азотистых веществ методом Кьельдаля. Навеску экстракта в количестве 0,8-1,2 г в колбе Кьельдаля заливают 20-30 мл серной кислоты (относительной плотностью 1,84) и ведут определение, как указано выше.
В качестве индикатора применяют смесь метиленового голубого (0,412 г) и метилового красного (0,624 г) в 500 мл 96%-ного этилового спирта.
Параллельно ставят контрольный опыт (без навески экстракта) и количество миллилитров 0,1 н. раствора NaOH, пошедшее на титрование контроля, вычитают из показателя, полученного при основном определении. Содержание азотистых веществ 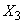 выражают в процентах в пересчете на сухое вещество экстракта и вычисляют, исходя из того, что 1 мл 0,1 н. HSO связывает 0,0014 г азота, по формуле
выражают в процентах в пересчете на сухое вещество экстракта и вычисляют, исходя из того, что 1 мл 0,1 н. HSO связывает 0,0014 г азота, по формуле
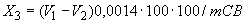 ,
,
где - количество 0,1 н. раствора серной кислоты, взятое в приемник, мл; - количество 0,1 н. раствора щелочи, пошедшее на титрование избытка серной кислоты, мл; - навеска экстракта, г; - содержание сухих веществ в экстракте, %.
Определение содержания сернистого газа. Сущность метода заключается в окислении свободного сернистого газа раствором йода. 50 мл раствора, приготовленного для определения кислотности (соответствует 0,5 г экстракта), помещают в колбу Эрленмейера, прибавляют 3-5 капель раствора 1%-ного крахмала и титруют 0,02 н. раствором йода в йодистом калии до появления синего окрашивания. Затем добавляют 2-3 капли разбавленной соляной кислоты (1:5), 7-10 капель 1%ного раствора крахмала и снова титруют до появления синего окрашивания, не исчезающего в течение 1 мин.
Содержание свободного сернистого ангидрида 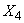 (в %) в пересчете на сухое вещество рассчитывают по формуле
(в %) в пересчете на сухое вещество рассчитывают по формуле
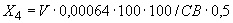 ,
,
где - количество 0,02 н. раствора йода, пошедшее на титрование, мл; - содержание сухих веществ в экстракте, %; 0,0064 - количество SO, соответствующее 1 мл 0,02 н. раствора йода, г; 0,5 - масса экстракта в 50 мл раствора, г.
Определение содержания общей золы. Сущность метода заключается в определении остатка несгоревшего экстракта при сжигании в муфельной печи при температуре 600-700 °С.
В предварительно прокаленном и взвешенном тигле взвешивают на аналитических весах 4-3 г экстракта, смачивают его поверхность 2-3 каплями чистого растительного масла (подсолнечного по ГОСТ 1129-73 или кукурузного по ГОСТ 8808-73), осторожно упаривают и сжигают на слабом огне. Затем тигель помещают для предварительного прокаливания в раскаленный муфель при температуре 400-500 °С, после чего поднимают температуру в муфеле до 600-700 °С. Тигель с золой прокаливают до постоянной массы. Содержание золы  (в %) в пересчете на сухое вещество экстракта определяют по формуле
(в %) в пересчете на сухое вещество экстракта определяют по формуле
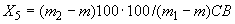 ,
,
где - масса тигля, г; - масса тигля с экстрактом, г; - масса тигля с золой, г; - содержание сухих веществ в экстракте, %.
Определение содержания биотина. В кукурузном экстракте количество биотина определяют по той же методике, что и в мелассе (см. с.47). Разница заключается в том, что навеска экстракта значительно меньше (для определения биотина берут от 0,01 до 0,03 г экстракта).
В кукурузном экстракте содержится биотина от 0,5 до 2 мкг в 1 г. В экстракте хорошего качества содержится от 1,6 до 2 мкг биотина в 1 г вещества.
Оценка кукурузного экстракта как источника стимуляторов роста дрожжей по технологической пробе. Метод основан на определении способности исследуемого образца увеличивать выход дрожжей. Хороший экстракт позволяет повысить выход дрожжей на 20-25% по сравнению с контролем, экстракт среднего качества - на 10-15%, неудовлетворительного - на 3-5%.
Дрожжи выращивают по схеме, приведенной для технологической оценки пригодности мелассы для дрожжевого производства (см. с.81).
При проведении анализа используют в обоих случаях одну и ту же мелассу в одинаковом количестве и одни и те же засевные дрожжи.
Исследуемый экстракт в количестве 9 г (6% к перерабатываемой мелассе) вносят в один из опытных сосудов со средой. Качество экстракта оценивают на основании полученной разницы в скорости роста дрожжей в опытном и контрольном аппаратах. Чем выше скорость роста дрожжей в результате добавления кукурузного экстракта, тем лучше его качество как стимулятора роста.
Ортофосфорная кислота
В качестве источника фосфорного питания используется термическая (пищевая) ортофосфорная кислота НРО (ГОСТ 10678-76). Пищевая ортофосфорная кислота марки А - бесцветная жидкость, прозрачная в слое 15-20 мм при рассматривании на белом фоне, содержащая кислоты не менее 73%, мышьяка не более 0,0003%.
Определяется содержание ортофосфорной кислоты, мышьяка.
Определение массовой доли ортофосфорной кислоты. Отбирают около 1 мл кислоты, разбавляют 50 мл дистиллированной воды и титруют 1 н. раствором щелочи, прибавив 3 капли метилового оранжевого.
1 мл 1 н. раствора гидроксида калия соответствует 0,09806 г НРО.
Содержание ортофосфорной кислоты (в %) вычисляют по формуле
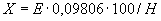 ,
,
где - количество 1 н. раствора щелочи, пошедшее на титрование, мл; - навеска кислоты, г.
Определение мышьяка в пищевой кислоте. Отвешивают 7 г с точностью 0,0001 г кислоты и вносят в сосуд прибора (см. рис.23), прибавляют 20 мл 0,1 н. раствора перманганата и 7 мл 25%-ного раствора серной кислоты. После двухчасового стояния прибавляют 6 мл 10%-ного раствора сернокислого железа и 7 мл 25%-ного раствора серной кислоты. После обесцвечивания и охлаждения раствора поступают так же, как при определении мышьяка в серной кислоте.
Карбамид
Карбамид (мочевина) используется как источник азота. Он представляет собой белые или слабоокрашенные кристаллы или гранулы, содержащие не менее 46% азота по ГОСТ 2081-75*.
______________
* На территории Российской Федерации действует ГОСТ 2081-92.**;
** На территории Российской Федерации с 01.03.2011 действует ГОСТ 2081-2010. - Примечание изготовителя базы данных.
Массовую долю азота в пересчете на сухое вещество определяют по ГОСТ 20851.1-75, разделу 2 со следующим дополнением: масса навески карбамида должна составлять 1 г, при этом количество серной кислоты должно быть 5 см, формалина - 40 см, титрование проводят 1 н. раствором гидроксида натрия.
Определение содержания сернокислого аммония и диаммонийфосфата в растворах
В дрожжевом производстве готовят 10, 15 и 20%-ные растворы питательных солей (сернокислого аммония или диаммонийфосфата). После каждого приготовления в них определяют концентрацию сухих веществ, по количеству которых можно контролировать содержание сернокислого аммония или диаммонийфосфата в растворе.
Содержание сернокислого аммония и диаммонийфосфата в растворе в процентах можно контролировать путем определения концентрации сухих веществ сахарометром или рефрактометром.
Количество сухих веществ в растворах сернокислого аммония и диаммонийфосфата разной концентрации представлено в табл.11
Таблица 11
|
Раствор |
Концентрация раствора, % |
Содержание СВ, % |
|
|
по сахарометру |
по рефрактометру |
||
|
Сернокислый аммоний |
10 |
13,8 |
10,6 |
|
15 |
19,7 |
14,6 |
|
|
20 |
24,8 |
18,7 |
|
|
Диаммонийфосфат |
10 |
15,5 |
12,1 |
|
15 |
21,7 |
17,7 |
|
|
20 |
26,5 |
21,6 |
|
Определение азота в растворах сернокислого аммония. Для определения берут 0,5-1 мл раствора в мерную колбу вместимостью 500 мл, в нее добавляют 400 мл воды и 100 мл 33%-ного раствора гидроксида натрия. Затем отгоняют аммиак и ведут расчеты, как это принято при определении азота по Кьельдалю.
Определение азота и фосфора в растворе диаммонийфосфата. Для определения аммиака в растворе диаммонийфосфата берут 1-2 мл раствора, переносят в коническую колбу вместимостью 150 мл, прибавляют 2-3 капли фенолфталеина и нейтрализуют 0,1 н. раствором гидроксида калия до слабо-розового окрашивания. Затем добавляют 10-15 мл нейтрализованного раствора формалина (40%-ного) и вторично титруют 0,1 н. раствором гидроксида калия до появления слабо-розового окрашивания.
Содержание аммиака (в %) определяют по формуле
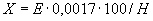 ,
,
где - количество 0,1 н. раствора гидроксида калия, пошедшее на титрование, мл; 0,0017 - количество NH, соответствующее 1 мл 0,1 н. раствора гидроксида калия; - количество диаммонийфосфата, содержащееся в растворе, взятом на определение, г.
Для определения фосфора в растворах диаммонийфосфата берут 1 мл раствора, разводят в мерной колбе на 500 или 1000 мл в зависимости от концентрации раствора. Затем 1 мл приготовленного раствора берут в пробирку для определения фосфора по методу Бригса.
АНАЛИЗ ВСПОМОГАТЕЛЬНЫХ МАТЕРИАЛОВ
Серная кислота
Серную кислоту применяют для подкисления мелассы при осветлении ее и для регулирования рН среды при выращивании дрожжей. Кроме того, ее используют для обработки засевных дрожжей в целях очистки их от бактериальной инфекции. В дрожжевом производстве применяют кислоту аккумуляторную (ГОСТ 667-73) и техническую (контактную улучшенную марок А и Б) (ГОСТ 2184-77) с содержанием серной кислоты 94,0%, мышьяка не более 0,00005-0,0001%.
Определяется содержание серной кислоты и мышьяка.
Определение массовой доли серной кислоты. Для определения содержания серной кислоты по относительной плотности в среднюю пробу серной кислоты, приведенную к температуре 20 °С, опускают денсиметр и отмечают показания, а затем находят процентное содержание серной кислоты по таблице.
Определение содержания серной кислоты титрованием: отвешивают 4-5 г исследуемой кислоты, крышка бюксы во время взвешивания должна быть закрыта. Затем количественно через воронку переводят кислоту в мерную колбу вместимостью 250 мл, в которую предварительно наливают около 150 мл дистиллированной воды. Бюксу нескольку раз тщательно смывают водой через воронку в мерную колбу, которую затем доливают водой до метки, и содержимое хорошо перемешивают. Из полученного таким образом раствора отбирают пипеткой 50 мл и титруют 0,5 н. раствором NaOH до слабо-розового окрашивания в присутствии метилового красного до перехода красной окраски раствора в желтую.
Массовая доля серной кислоты (в %) вычисляют по формуле
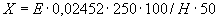 ,
,
где - количество 0,5 н. раствора гидроксида натрия, пошедшее на титрование, мл; - навеска испытуемой кислоты, г; 0,02452 количество HSO, соответствующее 1 мл 0,5 н. раствора гидроксида натрия.
Определение содержания мышьяка. Оно основано на свойствах всех мышьяковых соединений в кислом растворе восстанавливаться металлическим цинком в мышьяковистый водород.
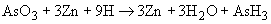 ;
;
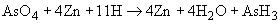 .
.
Эти реакции очень чувствительны, поэтому применяемые реактивы, особенно металлический цинк и серная кислота, должны быть абсолютно свободными от мышьяка. Полоски бумаги, пропитанные бромной ртутью, окрашиваются выделяющимся мышьяковистым водородом: чем больше мышьяка, тем больше пятно.
В серной кислоте, пригодной для дрожжевого производства, обнаруживаются следы мышьяка, величина пятна должна быть менее 2 мм, т.е. в кислоте не должно содержаться мышьяка более 0,0001%.
При действии AsH на HgBr сначала образуется As(HgBr), а затем при дальнейшем действии мышьяковистого водорода образуются соединения AsH(HgBr) и AsH.
Прибор (рис.23), в котором ведут определение, состоит из сосуда 1 вместимостью около 100 мл, трубки-насадки 2 (с внутренним диаметром около 7 мм и длиной около 70 мм), пришлифованной к горлу сосуда, трубки-уловителя 3 (длиной около 40 мм с внутренним диаметром около 4 мм), хорошо пришлифованной к верхнему концу трубки-насадки.
Рис.23. Прибор для определения мышьяка
При сборке аппарата можно пользоваться и обыкновенной конической колбой вместимостью 100 мл, причем отдельные части аппарата соединяют с помощью корковых пробок со вставленной вниз трубкой Алена указанных выше размеров. Перед использованием прибора в верхнюю часть насадки 2
помещают разрыхленный комочек ваты , смоченный раствором уксуснокислого свинца и отжатый между листами фильтровальной бумаги. Ниже вставляют полоски фильтровальной бумаги или марли , смоченной тем же раствором уксуснокислого свинца и отжатой между листами фильтровальной бумаги. Снизу трубку закрывают сухим разрыхленным комочком ваты . Для фиксации мышьяка в трубку-уловитель 3
помещают полоску бромортутной бумаги .
При проведении анализа в сосуд 1 наливают 50 мл дистиллированной воды, 2-3 капли 10%-ного раствора двухлористого олова и 1 или 2 мл испытуемой кислоты в зависимости от предполагаемого содержания мышьяка. После этого к раствору прибавляют соответственно 3 или 4 мл химически чистой серной кислоты без мышьяка. Затем прибор охлаждают, после этого в прибор бросают около 4 г химически чистого цинка, не содержащего мышьяка, и плотно вставляют заряженную указанным выше способом насадку 2 , а потом трубку-уловитель 3 с вложенной в него бромортутной бумажкой. Насадку закрывают колпачком из темной бумаги и весь прибор ставят в темное место.
Если в материале содержится мышьяк, то бромортутная бумажка окрашивается в зависимости от количества мышьяка в желтый цвет с оттенками до темно-бурого.
Реакцию считают законченной, когда растворится весь цинк. После этого прибор оставляют еще на 30 мин, затем вынимают полоску бромортутной бумаги из насадки 2 и определяют длину пятна*.
________________
* При определении мышьяка можно использовать прибор для определения сульфидов и сульфитов (см. рис.21). При этом в нижний конец трубки помешают кусочек чистой ваты, на которую кладут вату, смоченную уксуснокислым свинцом. Все плотно утрамбовывают стеклянной палочкой. Затем помещают полоску бромортутной бумаги. В верхний конец трубки помещают чистый кусочек ваты.
При использовании бумажных полосок шириной 2,5 мм соотношение между длиной пятен и содержанием мышьяка выражается следующими величинами:
|
Массовая доля мышьяка, мг |
0,05 |
0,035 |
0,025 |
0,015 |
0,05 |
0,0035 |
0,0025 |
0,0015 |
|
Длина пятна, мм |
30 |
26 |
18 |
10 |
6 |
4 |
3 |
2 |
Если при определении массовой доли мышьяка окраска полосок выходит за пределы окраски шкалы, то для испытания берут большее или меньшее количество кислоты.
Массовая доля мышьяка (в %) определяется по формуле
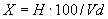 ,
,
где - количество мышьяка, содержащееся в испытуемой кислоте и определяемое сравнением окраски бромортутной бумажки, г; - количество испытуемой кислоты, мл;  - относительная плотность испытуемой кислоты.
- относительная плотность испытуемой кислоты.
Пример. На анализ взято 5 мл испытуемой кислоты, относительная плотность серной кислоты 1,84, длина полученного пятна 5 мм, что соответствует 0,0005 мг или 0,000005 г мышьяка. Подставляем полученную величину в формулу и получаем массовую долю мышьяка в исследуемом веществе (в %)
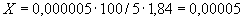 %.
%.
Хлористый калий (технический)
Хлористый калий используют как источник калия в случае недостаточного содержания его в мелассе. Следует применять калий I и II сортов, содержащий хлористого калия не менее 95-98%, влаги не более 1%.
Пробу для анализа отбирают при помощи щупа не менее чем из 10 мест, общая масса средней пробы должна быть не менее 5 кг.
Определяется содержание влаги и хлористого калия.
Определение массовой доли влаги. Отвешивают около 10 г испытуемого вещества и сушат до постоянной массы в сушильном шкафу при температуре 105-110 °С.
Содержание влаги (в %) вычисляют по формуле
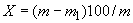 ,
,
где - масса хлористого калия до высушивания, г; - масса хлористого калия после высушивания, г.
Определение содержания хлористого калия. Для проведения анализа берут 5 г хлористого калия, взвешивают с погрешностью не более 0,0002 г, переносят в стакан вместимостью 250 мл, добавляют 100-150 мл воды и нагревают до кипения. Раствор фильтруют в мерную колбу вместимостью 500 мл через фильтр "белая лента" диаметром 9 см. Фильтр с нерастворимым в воде остатком промывают водой до отрицательной реакции на ион хлора (проба с раствором азотнокислого серебра), объем раствора доводят водой до метки и тщательно перемешивают.
Из колбы пипеткой отбирают 50 мл раствора в мерную колбу вместимостью 500 мл, доводят объем раствора водой до метки и тщательно перемешивают.
50 мл раствора после второго разбавления отбирают пипеткой в стакан вместимостью 100 мл, прибавляют 1-2 капли индикатора (метиленовый красный) и 10%-ный раствор уксусной кислоты до изменения цвета раствора, нагревают на водяной бане до 40 °С и осаждают тетрафенилборат калия, приливая по каплям при перемешивании 10 мл 3,5%-ного раствора тетрафенилбората натрия.
Раствору с осадком дают отстояться на бане в течение 5 мин, охлаждают в проточной воде до комнатной температуры и осадок отфильтровывают через высушенный до постоянной массы фильтрующий тигель типа ТФ ПОР16. Осадок из стакана переносят на фильтр и промывают небольшими порциями (3-4 мл) промывного раствора, каждый раз отстаивая раствор досуха. Затем осадок промывают три раза по 5 мл холодной водой. Общий расход промывных вод не должен превышать 50 мл. Фильтр с осадком сушат в сушильном шкафу при 120 °С до постоянной массы.
Содержание хлористого калия (в %) вычисляют по формуле
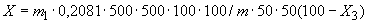 ,
,
где - масса осадка тетрафенилбората калия, г; 0,2081 - коэффициент пересчета K[B(CH)] на KCl; - навеска, г; - содержание влаги, %.
Содержание окиси калия 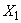 (в %) определяют по формуле
(в %) определяют по формуле
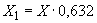 ,
,
где - содержание хлористого калия, %; 0,632 - коэффициент пересчета хлористого калия на окись калия.
Углекислый калий (поташ)
Поташ используют как источник калия при недостатке его в мелассе и для регулирования рН среды в дрожжерастильных аппаратах. Он представляет собой порошок белого цвета, легко растворимый в воде, раствор имеет щелочную реакцию.
Выпускают поташ трех сортов по действующему ГОСТу. Для производственных целей используют поташ I сорта, содержащий углекислого калия не менее 98% (ГОСТ 10690-73). Определяется содержание углекислого калия.
Определение массовой доли углекислого калия. Около 2 г углекислого калия взвешивают с погрешностью до 0,0002 г, помещают в коническую колбу вместимостью 250 мл, приливают 50 мл воды, нагревают до кипения, прибавляют 2-3 капли раствора метилового красного и титруют раствором соляной кислоты до перехода желтой окраски в малиновую.
Содержание углекислого калия (в %) вычисляют по формуле
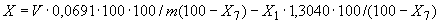 ,
,
где - навеска углекислого калия, г; - количество 1 н. раствора соляной кислоты, израсходованное на титрование, мл; - содержание натрия в пересчете на NaCO, определенное по п.3.3, ГОСТ 10690-73, %; 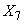 - потери в массе при промывании, определенные по п.3.9, ГОСТ 10690-73, %; 0,0691 - количество углекислого калия, соответствующее 1 мл 1 н. раствора соляной кислоты, г; 1,3040 - коэффициент пересчета углекислого натрия на углекислый калий.
- потери в массе при промывании, определенные по п.3.9, ГОСТ 10690-73, %; 0,0691 - количество углекислого калия, соответствующее 1 мл 1 н. раствора соляной кислоты, г; 1,3040 - коэффициент пересчета углекислого натрия на углекислый калий.
Магний сернокислый
Сернокислый магний используют как источник магния при недостатке его в мелассе и производственной воде. Сернокислый магний (ГОСТ 4523-77) представляет собой белый кристаллический порошок, растворимый в воде.
Содержание MgSО·7НО в реактиве должно быть не менее 99%. Содержание мышьяка в препарате допускается не более 0,00004%.
АНАЛИЗ ДЕЗИНФИЦИРУЮЩИХ И МОЮЩИХ СРЕДСТВ
Хлорная известь
Хлорную известь получают при насыщении гашеной извести (пушонки) газообразным хлором. Применяется для дезинфекции аппаратуры и помещений завода. Определяется количество активного хлора.
Ход определения заключается в следующем. Навеску около 10 г растирают в ступке в небольшом количестве дистиллированной воды, переносят в колбу вместимостью 1 л, доливают водой до метки, тщательно перемешивают, отбирают пипеткой 50 мл раствора, прибавляют 1-2 г йодистого калия и 10-20 капель концентрированной соляной кислоты. Выделившийся йод титруют 0,1 н. раствором гипосульфита до тех пор, пока окраска жидкости из коричневой не превратится в светло-желтую. Добавляют 1-2 капли раствора крахмала и дотитровывают содержимое колбы гипосульфитом до исчезновения голубого окрашивания.
Содержание активного хлора (в %) рассчитывают по формуле
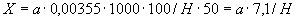 ,
,
где - количество 0,1 н. раствора гипосульфита, израсходованное на титрование, мл; - навеска хлорной извести, г; 0,00355 - количество хлора, соответствующее 1 мл 1 н. раствора гипосульфита, г.
Пример. Навеска хлорной извести 9,9781 г, на титрование 50 мл раствора затрачено 41,5 мл раствора гипосульфита. Содержание активного хлора (в %)
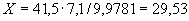 .
.
Каустическая сода (гидроксид натрия,
ГОСТ 2263-79)
Каустическая сода применяется для мойки и дезинфекции аппаратуры и трубопроводов. Определяется массовая доля гидроксида натрия и углекислого натрия.
Ход определения заключается в следующем. 100 г твердого или 200 г жидкого продукта растворяют в дистиллированной воде, не содержащей диоксида углерода, и доводят объем до 500 мл в мерной колбе. В две конические колбы отмеривают пипеткой по 5 мл приготовленного раствора.
1. Определяют содержание NaOH. В одну колбу добавляют 5 мл 10%-ного раствора хлористого бария, оставляют в покое на 15 мин, после чего содержимое этой колбы титруют 1 н. раствором соляной кислоты в присутствии фенолфталеина до обесцвечивания раствора.
Содержание гидроокиси натрия (в %) определяют по формуле
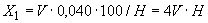 ,
,
где - содержание гидроксида натрия, %; - количество 1 н. раствора соляной кислоты, затраченное на титрование содержимого первой колбы, мл; 0,040 - количество гидроксида натрия, соответствующее 1 мл 1 н. раствора кислоты, г; - навеска, соответствующая 5 мл приготовленного раствора, т.е. 1 г сухого исходного материала или 2 г исходного жидкого материала, г.
2. Определяют содержание углекислого натрия. Содержимое 2-й колбы титруют 1 н. раствором соляной кислоты в присутствии метилового оранжевого до розового окрашивания раствора.
Содержание NaCO вычисляется по формуле
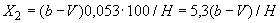 ,
,
где 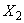 - содержание углекислого натрия, %; - количество 1 н. раствора соляной кислоты, затраченное на титрование содержимого второй колбы, мл; - количество 1 н. раствора соляной кислоты, затраченное на титрование содержимого первой колбы, мл; - навеска, соответствующая 5 мл приготовленного раствора, т.е. 1 г сухого исходного материала или 2 г исходного жидкого материала, г; 0,053 - количество углекислой соды, соответствующее 1 мл 1 н. раствора кислоты, г.
- содержание углекислого натрия, %; - количество 1 н. раствора соляной кислоты, затраченное на титрование содержимого второй колбы, мл; - количество 1 н. раствора соляной кислоты, затраченное на титрование содержимого первой колбы, мл; - навеска, соответствующая 5 мл приготовленного раствора, т.е. 1 г сухого исходного материала или 2 г исходного жидкого материала, г; 0,053 - количество углекислой соды, соответствующее 1 мл 1 н. раствора кислоты, г.
Пример. Анализируется жидкий каустик. На титрование содержимого 1-й колбы затрачено 20,6 мл 1 н. раствора кислоты, на титрование содержимого 2-й колбы - 23,6 мл 1 н. кислоты.
Содержание NaOH в жидком каустике (в %) будет 4,0-20,6/2=41,2.
Содержание NaCO (в %) составит 5,3(23,6-20,6)/2=7,95.
Формалин технический (ГОСТ 1625-75*)
______________
* На территории Российской Федерации действует ГОСТ 1625-89. - Примечание изготовителя базы данных.
Формалин - раствор формальдегида в воде. Его применяют для дезинфекции трубопроводов в виде 0,5-1%-ных растворов. Для окуривания дрожжерастильных аппаратов газообразным формальдегидом используют реакцию взаимодействия формалина с марганцевокислым калием, при этом выделяется свободный формальдегид в виде газа. Определяется содержание формальдегида.
Для определения массовой доли формальдегида 3-3,5 г формалина взвешивают в колбе с пробкой с погрешностью не более 0,0002 г, приливают 10 мл дистиллированной воды, 2 капли тимолфталеина и нейтрализуют гидроксидом натрия до бледно-голубой окраски.
В другую колбу помещают 75 мл раствора сульфита натрия, добавляют 2 капли тимолфталеина и нейтрализуют 0,1 н. раствором соляной кислоты до исчезновения голубой окраски или раствором гидроксида натрия до бледно-голубой окраски.
Нейтральный раствор сульфита натрия переливают в колбу с навеской, перемешивают в течение 2 мин и титруют 1 н. раствором соляной кислоты до исчезновения голубой окраски.
Содержание формальдегида в формалине (в %) вычисляют по формуле
 ,
,
где - количество 1 н. раствора соляной кислоты, израсходованное на титрование выделившегося гидроксида натрия, мл; 0,03003 - количество формальдегида, соответствующее 1 мл 1 н. раствора соляной кислоты, г; - масса анализируемой пробы, г.
Известь строительная ( ГОСТ 9179-77)
Известь свежегашеную или в виде известкового молока используют для дезинфекции помещений. В свежегашеной извести может содержаться до 85% СаО. Определяют содержание СаО.
Взвешивают около 1 г извести с точностью до 0,001 г. Навеску без потерь переносят в коническую колбу и наливают 25 мл горячей дистиллированной воды. Колбу закрывают резиновой пробкой и оставляют на 30 мин, после чего, добавив 2-3 капли раствора фенолфталеина, титруют содержимое кислотой до стойкого исчезновения розового окрашивания.
Количество окиси кальция (в %) рассчитывают по формуле
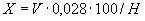 ,
,
где - количество 0,1 н. раствора кислоты, затраченное на титрование, мл; - навеска извести, г; 0,028 - количество окиси кальция, соответствующее 1 мл 0,1 н. раствора кислоты, г.
Пример. Навеска извести, взятая на определение, 0,9764 г; на титрование израсходовано 29,2 мл 0,1 н. серной кислоты.
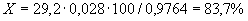 .
.
АНАЛИЗ ПЕНОГАСИТЕЛЕЙ
В процессе выращивания дрожжей в дрожжерастильных аппаратах образуется пена, для гашения которой применяют пеногасители. В дрожжевом производстве обычно для гашения пены применяют техническую олеиновую кислоту марок А и В (ГОСТ 7580-55*) с содержанием жирных кислот 95% и температурой застывания 10-16 °С. Кроме того, используют техническую олеиновую кислоту марки О, дистиллированные жирные кислоты подсолнечного и соевого масел, смесь дистиллированных жирных кислот оливкового и горчичного масел, масло хлопковое и подсолнечное и хлебопекарный фосфатидный концентрат. Определяются температура застывания, йодное и кислотное числа.
______________
* На территории Российской Федерации действует ГОСТ 7580-91. - Примечание изготовителя базы данных.
Определение температуры застывания. В пробирку диаметром 30 мм отмеривают 25-30 мл технической олеиновой кислоты. Пробирку помещают в стеклянный сосуд, охлаждаемый смесью льда с водой.
В пробирку помещают термометр с ценой деления 0,1 °С так, чтобы он не касался стенок пробирки, и помешивают им олеин до появления мути, после чего дают олеину застыть и отмечают показания термометра.
Температурой застывания считают ту температуру, на которой остановится ртуть термометра, или ту максимальную температуру, до которой поднимается ртуть, если после ее понижения будет замечено вновь некоторое повышение температуры.
Определение массовой доли жира. Взвешивают 10 г пеногасителя с точностью 0,0002 г и добавляют 100 мл этилового эфира. Колбу закрывают пробкой, в течение 2 ч периодически встряхивают (через 30 мин).
После отстаивания в течение нескольких часов в предварительно взвешенную коническую колбу отбирают 50 мл прозрачной вытяжки. Эфир отгоняют в вытяжном шкафу на водяной бане и колбу ставят в сушильный шкаф на 30 мин при температуре 100 °С. После охлаждения в эксикаторе масло, выделенное из 5 г исследуемого жира, взвешивают и, умножая массу на 20, получают содержание жира в процентах.
Определение йодного числа. Йодное число - это количество граммов йода, присоединившееся к 100 г исследуемого вещества. Это число характеризует степень непредельности жира или жирных кислот.
Определение йодного числа позволяет также установить природу жирной кислоты, так как, например, йодное число олеиновой кислоты равно 89,3, а линолевой - 181,16. Такая значительная разница между йодными числами этих кислот не только позволяет определить, какая из двух кислот имеется, но и дает возможность установить их количественное соотношение, если известно йодное число смеси.
Характеристика различных жиров и жирных кислот по величине йодного числа приведена ниже.
|
Жирные кислоты |
|
|
олеиновая |
86-89,3 |
|
линолевая |
181,16 |
|
Масло |
|
|
подсолнечное |
125-135 |
|
хлопковое |
101-117 |
|
льняное |
168-176 |
|
конопляное |
140-158 |
|
репейное |
94-106 |
Ход определения заключается в следующем. В колбу с притертой пробкой вносят около 0,2-0,3 г олеина, взвешенного с точностью 0,0002 г, прибавляют 10 мл хлороформа и 25 мл йодно-ртутного раствора. Колбу закрывают пробкой, смоченной раствором йодистого калия. Раствор олеина и йодно-ртутную смесь смешивают, после чего раствор должен быть совершенно прозрачный, в противном случае к смеси добавляют еще некоторое количество хлороформа. Одновременно с первой колбой ставят вторую (контрольную) колбу при тех же условиях, т.е. с 10 мл хлороформа и 25 мл йодно-ртутного раствора. В случае добавления хлороформа в колбу с навеской олеина вносят хлороформ и во вторую (контрольную) колбу. Обе колбы оставляют в темноте при температуре 20 °С на 12-18 ч.
По окончании настаивания в колбу с испытуемым олеином приливают 20 мл раствора йодистого калия. В случае выпадения красного осадка йодистой ртути прибавляют еще раствора йодистого калия до растворения выпавшего осадка. Такое же количество раствора йодистого калия прибавляют и в контрольную пробу. После этого в обе колбы прибавляют по 100 мл воды.
Содержимое обеих колб титруют 0,1 н. раствором гипосульфита до появления желтого окрашивания. После этого прибавляют 1 мл раствора крахмала и продолжают титрование до исчезновения синей окраски.
Йодное число вычисляют по формуле
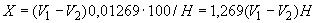 ,
,
где - навеска олеина, г; - количество 0,1 н. раствора гипосульфита, пошедшее на титрование контрольной пробы, мл; - количество 0,1 н. раствора гипосульфита, пошедшее на титрование пробы с навеской олеина, мл; 0,01269 - количество йода, соответствующее 1 мл 0,1 н. раствора гипосульфита, г.
Определение кислотного числа. Кислотное число характеризует количество свободных жирных кислот в пеногасителе и выражается в миллиграммах гидроксида калия, необходимого для нейтрализации свободных кислот, входящих в состав 1 г исследуемого вещества.
Взвешивают около 1-2 г олеина с точностью 0,0002 г, растворяют в 20 мл нейтрального, нагретого до кипения этилового спирта. После полного растворения олеина при нагревании до начала кипения раствор титруют 0,5 н. спиртовым раствором гидроксида калия до слабо-розового окрашивания (индикатор фенолфталеин).
Количество миллиграммов гидроксида калия, израсходованное на нейтрализацию 1 г олеина, определяет кислотное число.
Кислотное число вычисляют по формуле
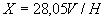 ,
,
где - навеска олеина, г; - количество гидроксида калия, пошедшее на титрование, мл; 28,05 - количество гидроксида калия, содержащееся в 1 мл 0,5 н. раствора гидроокиси калия, мг.
АНАЛИЗ КУЛЬТУРАЛЬНОЙ СРЕДЫ
В процессе выращивания дрожжей в культуральной среде определяют температуру, рН (активную кислотность), концентрацию сухих веществ, концентрацию дрожжей, остаточные питательные вещества (усваиваемый азот, сахар, спирт), а также растворенный кислород, скорость роста дрожжей и наличие нитритов.
Температура, рН, концентрация сухих веществ и концентрация дрожжей в среде характеризуют правильность ведения процесса в соответствии с заданной технологией. Содержание усваиваемого азота указывает на сбалансированность азотного питания, а сахара - углеводного питания и степени аэрации. По содержанию спирта и растворенного кислорода в среде обычно судят об интенсивности аэрирования. Наличие нитритов в среде нежелательно, так как при этом понижается выход дрожжей.
Определение температуры
Температуру определяют либо вручную, либо автоматически, например лагометрами, потенциометрами с термометрами сопротивления. В первом случае из дрожжерастильного аппарата отбирают пробу не менее 1 л и, погрузив тотчас термометр, определяют температуру. Во втором случае температура регистрируется на шкале прибора.
Определение рН
Активную кислотность измеряют специальными приборами, называемыми рН-метрами. Из дрожжерастильного аппарата отбирают пробу (не менее 100 мл), помещают ее в стаканчик рН-метра, устанавливают диапазон измерения температуры культуральной среды и по шкале прибора определяют рН в соответствии с инструкцией, приложенной к прибору заводом-изготовителем.
С помощью рН-метра осуществляется и автоматический контроль среды, при этом по шкале прибора отмечается величина этого показателя.
Определение концентрации дрожжей
Концентрацию дрожжей в культуральной среде определяют либо путем выделения на фильтрах, либо по мутности среды с помощью приборов, называемых нефелометрами.
В первом случае из дрожжерастильного аппарата отбирают 1 л среды, отфильтровывают на воронке Бюхнера через двойной фильтр крупнопористой бумаги. Фильтрование заканчивают при получении пласта прессованных дрожжей плотной консистенции. Дрожжи взвешивают на технических весах, не снимая их с фильтровальной бумаги, причем нижний кружок фильтровальной бумаги помещают на чашку весов с гирями, чтобы избежать потери дрожжей. Взвесив пласт дрожжей, определяют влажность и вносят коррективу в массу дрожжей, чтобы установить концентрацию прессованных дрожжей влажностью 75%.
Пример 1. Масса пластинки дрожжей 60 г/л, влажность 72%. При этом концентрация дрожжей в пересчете на 25% СВ составит
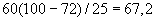 г/л.
г/л.
Для оперативности анализа можно пользоваться экспериментально установленным для данной установки коэффициентом, который обычно составляет 0,8-0,9. В данном случае для определения концентрации дрожжей массу пластинки отфильтрованных на воронке Бюхнера дрожжей умножают на установленный коэффициент.
Пример 2. Масса пластинки отфильтрованных дрожжей составляет 75 г/л, а коэффициент пересчета для данной установки - 0,8. Концентрация дрожжей при этом будет
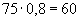 г/л.
г/л.
Определение концентрации дрожжей с помощью нефелометра см. определение содержания биотина. Общее количество дрожжей, находящихся в данный момент в дрожжерастильном аппарате, определяют путем умножения концентрации дрожжей на объем среды.
Пример 3. При объеме среды 60 м и концентрации дрожжей 67,2 г/л (или 67,2 кг/м) в дрожжерастильном аппарате содержится дрожжей
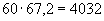 кг.
кг.
Метод определения содержания усваиваемого азота
Усваиваемый азот в среде обычно определяют формольным титрованием. Сущность метода состоит в том, что формалин связывает аминогруппы большинства аминокислот, освобождая при этом карбоксильные группы, CHNHCOOH+НСО=CHN=СНСООН+НО, которые затем оттитровывают щелочью CHN=СНСООН+NaOH=CHN=CHCOONa+НО, и косвенным путем определяют количество усваиваемого азота (аминных групп).
К 10 мл отфильтрованной от дрожжей бражки добавляют 90 мл дистиллированной воды и титруют скачала 0,1 н. раствором гидроксида натрия с фенолфталеином до появления слабо-розового окрашивания, затем в эту же колбу добавляют 5 мл нейтрального 40%-ного формалина и титруют вторично тем же раствором гидроксида натрия до появления слабо-розового окрашивания (приготовление нейтрального раствора формалина см. в приложении II).
Количество миллилитров 0,1 н. раствора гидроксида натрия, пошедшее на вторичное титрование (или количество миллилитров нормальной щелочи на 100 мл среды), называют формольным числом. Эта величина относительная и характеризует количество растворенного в среде аммонийного и аминного азота, абсолютное количество которого в граммах можно вычислить следующим образом:
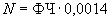 ,
,
где 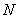 - количество усваиваемого азота, г;
- количество усваиваемого азота, г; 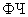 - формольное число, мл 0,1 н. раствор NaOH; 0,0014 - коэффициент, соответствующий содержанию азота в 1 мл 0,1 н. раствора NaOH, г.
- формольное число, мл 0,1 н. раствор NaOH; 0,0014 - коэффициент, соответствующий содержанию азота в 1 мл 0,1 н. раствора NaOH, г.
В период интенсивного роста дрожжей по воздушно-приточному или воздушно-проточному способу формольное число колеблется в пределах 0,7-1,5. При дозревании оно понижается до 0,1-0,3. Снижение формольного числа в период интенсивного роста до 0,5 указывает на несбалансированность азотного питания. При этом требуется подача дополнительного азотного питания.
Определение содержания остаточных сахаров
В процессе роста и размножения не все редуцирующие вещества, в том числе и сахара, используются клеткой. В культуральной среде обычно остается 0,1-0,05% неиспользованного сахара. Для определения остаточных сахаров предлагается ускоренный фотоколориметрический метод определения сахара в бражке с помощью хлористого трифенилтетразолийхлорида (ТТХ), разработанный в ЛО ВНИИХП, и эбулиостатический метод.
Фотоколориметрический метод. Этот метод не требует больших затрат времени, по точности не уступает методу Бертрана, прост в исполнении, применим для определения сахара в мутных и окрашенных жидкостях.
Принцип метода основан на восстановлении сахаром бесцветного трифенилтетразолийхлорида (ТТХ) с образованием красного формазона. Интенсивность окраски пропорциональна количеству сахара в пробе. Метод применим для растворов, содержащих от 50 до 500 мкг сахара в пробе.
Определение начинают с построения калибровочного графика. Готовят стандартный раствор, содержащий 500 мкг безводной глюкозы в 1 мл. Из него делают серию разведений (табл.12).
Таблица 12
|
Номер пробирки |
Количество основного раствора, мл |
Количество воды, мл |
Общий объем, мл |
Содержание глюкозы в пробе, мкг |
|
1 |
7,5 |
0 |
7,5 |
500 |
|
2 |
4,5 |
3 |
7,5 |
300 |
|
3 |
3,0 |
4,5 |
7,5 |
200 |
|
4 |
1,5 |
6,0 |
7,5 |
100 |
|
5 |
0,75 |
6,75 |
7,5 |
50 |
Из этих разведений берут по 1 мл в чистые пробирки, добавляют по 0,5 мл воды, 1 мл 1 н. NaOH и по 0,5 мл трифенилтетразолийхлорида и помещают пробирки в кипящую водяную баню на 3 мин. Этого времени достаточно для полного развития окраски. Пробирки быстро охлаждают в воде со льдом и сейчас же добавляют 3 мл бутанола. Пробирки энергично встряхивают, затем после расслоения жидкости из верхнего окрашенного слоя отбирают 0,5 мл и переносят в пробирку с 3 мл чистого бутанола. Пробу встряхивают для перемешивания ее содержимого и колориметрируют на фотоэлектрокалориметре с зеленым фильтром (длина волны 610-750 нм) в кюветах шириной 3 мм. В контрольную кювету наливают чистый бутанол.
По полученным точкам строят калибровочную кривую, откладывая на горизонтальной оси концентрацию раствора глюкозы в микрограммах, на вертикальной оси - величину экстинкции.
Ход определения заключается в следующем. Бражку отделяют от дрожжей на воронке Бюхнера небольшого размера или настольной центрифуге. Колба Бунзена и воронка Бюхнера должны быть совершенно чистыми и сухими. Так как мелассная среда содержит сахарозу, сначала проводят гидролиз этого дисахарида в 2%-ной серной кислоте. Для этого в чистую сухую пробирку вносят от 0,1 до 0,8 мл бражки (количество зависит от того, сколько ожидается сахара в среде в данный момент), доводят объем пробы до 0,8 мл дистиллированной водой (если берут на анализ 0,8 мл, воду не добавляют) и вносят 0,2 мл 10%-ной серной кислоты. Пробирку помещают в кипящую водяную баню на 5 мин. Охлаждают в воде со льдом, нейтрализуют 0,35 мл 5%-ной NaOH, доводят объем дистиллированной водой до 1,5 мл. Затем приливают 1 мл 1 н. раствора NaOH и 0,5 мл 0,5%-ного водяного раствора ТТХ, перемешивают, нагревают в кипящей водяной бане в течение 3 мин.
Затем пробу быстро охлаждают в воде со льдом и сейчас же добавляют 3 мл бутанола. Пробирку энергично встряхивают, затем после расслоения жидкости из верхнего окрашенного слоя отбирают 0,5 мл и переносят в пробирку с 3 мл чистого бутанола. Пробу встряхивают, чтобы перемешать ее содержимое, и колориметрируют на фотоэлектроколориметре с зеленым фильтром (длина волны 610-750 нм) в кюветах шириной 3 мм (в контрольной кювете находится чистый бутанол). По полученной величине экстинкции по калибровочной кривой находят содержание глюкозы в микрограммах. При определении сахарозы полученную величину следует умножить на коэффициент 0,95, чтобы получить содержание сахарозы.
Если определение ведут в начале дрожжерастильного процесса, когда сахара в среде много, бражку предварительно разводят так, чтобы содержание РВ в пробе не превышало 500 мкг в 0,1 мл. Тогда при расчете необходимо учесть и это разведение.
Пример. На анализ взяли 0,4 мл бражки. При колориметрии получена экстинкция 0,185. Это соответствует 152 мкг глюкозы или 152·0,95=144,4 мкг сахарозы.
Итак, в 0,4 мл содержится 144,4 мкг сахара, тогда в 1 мл - мкг.
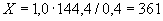 мкг.
мкг.
Если бражку разводили перед тем, как взять на анализ, данную цифру следует умножить на разведение.
Эбулиостатический метод (ускоренный).
Определение редуцирующих веществ (РВ) основано на реакции окисления их медно-щелочным раствором. При этом медь из двухвалентной (СuО) переходит в одновалентную (СuО). Во избежание выпадания CuО в осадок к медно-щелочному раствору прибавляют желтую кровяную соль, образующую прочное, хорошо растворимое соединение с одновалентной медью.
В эбулиостатическом методе использован принцип прямого титрования. Горячий медно-щелочный раствор (98 °С) титруют раствором сахара в специальном приборе - эбулиостате (рис.24), в котором обогрев реагирующей жидкости осуществляется пропусканием пара через реагирующую жидкость и поддерживается паровой рубашкой. Такой обогрев позволяет проводить анализ в потоке водяного пара без доступа воздуха при хорошем перемешивании и без местных перегревов. Индикатором является метиленовая синь, которая в окислительной среде имеет синюю окраску, а в восстановительной - бесцветна. О конце титрования судят по изменению цвета медно-щелочного раствора сахара.
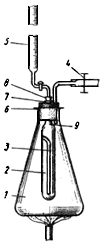
Рис.24. Эбулиостат
Эбулиостат состоит из двух сосудов: внешнего 1 (обычная коническая колба вместимостью 1 л) и внутреннего 2 , представляющего собой широкую пробирку, суженную вверху и имеющую внутри трубку 3 для пара, один конец которой впаян вверху боковой стенки сосуда 2 , а второй остается свободным и доходит почти до дна сосуда 2 . Верхнее отверстие эбулиостата 7 во время титрования закрывают пробкой 8 , для выхода пара имеется маленькое отверстие 9 на боковой стенке верхней узкой части эбулиостата.
Ход определения заключается в следующем. Во внешний сосуд 1 эбулиостата наливают воду и ставят его на нагревательный прибор. Когда вода закипит, вынимают внутренний сосуд 2 , который соединяется с сосудом 1 пробкой 6 ; наливают в него по 5 мл первого и второго растворов (см. приложение II), медленно вставляют его обратно в сосуд 1 с кипящей водой и закрывают верхнее отверстие эбулиостата пробкой 8 , надетой на кончик бюретки 5 с анализируемым раствором, содержащим сахар. Медно-щелочный раствор в сосуде 2 нагревается до 98 °С в течение 30-40 с, как только пар начнет проходить через медно-щелочный раствор со скоростью 3-4 пузырька в секунду. Если пар долго не проходит через внутренний сосуд 2 , надо уменьшить выход его из сосуда 1 через отводную трубку, подвинтив зажим 4 на трубке. При титровании следят за тем, чтобы вследствие очень быстрого приливания холодного раствора сахара не прекратилось прохождение пара через реагирующую жидкость в сосуде 2 . В случае прекращения видимого кипения жидкости в сосуде 2 уменьшают скорость приливания сахарного раствора из бюретки 5 .
Если концентрация сахара приблизительно известна, то приливают сначала около 80% того количества раствора, которое должно идти на титрование, затем ждут 2 мин (по песочным часам) и дотитровывают, прибавляя раствор сахара со скоростью 1 капля в 5-6 с. Увеличение скорости приливания последних капель приводит к заниженным результатам анализа. Появление желтой окраски жидкости после прибавления одной капли испытуемого раствора указывает на конец титрования.
Если концентрация сахара в анализируемой пробе неизвестна, то предварительно делают ориентировочное определение.
Концентрацию редуцирующих веществ в анализируемом растворе (в %) вычисляют по формуле
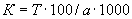 ,
,
где - условный титр медного раствора по глюкозе, т.е. количество глюкозы, которое восстанавливает 10 мл медно-щелочного раствора, мг; - количество раствора редуцирующих веществ, пошедшее на титрование, мл.
В жидкостях с концентрацией сахаров менее 0,05% рекомендуется применять обратный порядок титрования. Для этого 1-5 мл анализируемого раствора сахара приливают пипеткой в сосуд 2
непосредственно после добавления первого и второго растворов, а затем, как только пар начнет проходить через реагирующую жидкость в сосуде 2
, ждут 2 мин, после чего дотитровывают из бюретки раствором сахара известной концентрации со скоростью одна капля в 5-6 с до появления желтой или оранжевой окраски. Концентрацию редуцирующих веществ (в %) определяют по формуле
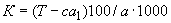 ,
,
где 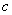 - количество редуцирующих веществ в 1 мл раствора сахара известной концентрации, мг; - количество раствора сахара известной концентрации, пошедшее на дотитрование, мл; - количество исследуемого раствора, мл.
- количество редуцирующих веществ в 1 мл раствора сахара известной концентрации, мг; - количество раствора сахара известной концентрации, пошедшее на дотитрование, мл; - количество исследуемого раствора, мл.
Определение содержания спирта (по Мартену)
Количество спирта в культуральной среде колеблется от 0,01 до 0,09%. Оно может составлять и значительно большие величины, например 1,5%, что зависит от степени аэрации культуральной среды и технологии получения дрожжей.
Спирт определяют по видоизмененному методу Мартена. Сущность метода заключается в окислении спирта смесью двухромовокислого калия с серной кислотой, при этом спирт окисляется до уксусной кислоты и воды.
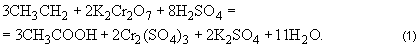
Избыток двухромовокислого калия (хромпика) оттитровывают раствором соли Мора.
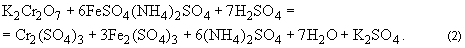
Конец реакции определяют по дифениламину или красной кровяной соли (раствору железосинеродистого калия).
Концентрацию спирта вычисляют по количеству двухромовокислого калия, пошедшему на окисление спирта.
Ход определения заключается в следующем. В низкогорлую круглодонную колбу вместимостью 50 мл (рис.25) наливают 10 мл профильтрованной среды и 0,5 мл 0,1 н. NaOH. В трехшариковый приемник вводят 25 мл раствора двухромовокислого калия (42,6087 г в 1 л), 5 мл серной кислоты относительной плотностью 1,84 и 5 мл ортофосфорной кислоты относительной плотностью 1,66. Приемник помещают в кристаллизатор со льдом. Колбу плотно закрывают резиновой пробкой с отводной трубкой длиной около 50 см, согнутой под углом 45° и суженой на конце. Суженый конец трубки помещают в трехшариковый приемник так, чтобы он почти касался дна приемника. После соединения колбы с приемником содержимое колбы осторожно нагревают и в течение 10-12 мин отгоняют 2/3 ее содержимого, следя за тем, чтобы образующаяся при нагревании пена не попадала в приемник.
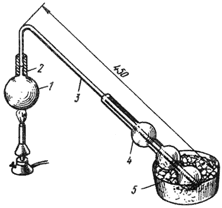
Рис.25. Прибор для определения спирта по Мартену:
1 - колба для отгона; 2 - соединительная пробка; 3 - отводная трубка; 4 - трехшариковый приемник; 5 - кристаллизатор со льдом
Жидкость из приемника переливают в колбу с широким горлом вместимостью 750 мл. Отводную трубку и приемник тщательно промывают, промывные воды присоединяют к основной жидкости и доводят объем ее дистиллированной водой приблизительно до 400 мл. После тщательного перемешивания полученную смесь оттитровывают раствором соли Мора с индикатором дифениламином, которого добавляют 5-6 капель. Переход окраски дифениламина в изумрудно-зеленый цвет определяет конец титрования.
Конец реакции можно определить также по раствору красной кровяной соли путем капельной пробы по появлению синего окрашивания от смешивания одной капли титруемой жидкости с одной каплей раствора красной кровяной соли. При этом виде титрования пробы следует брать после каждого прибавления 0,1-0,2 мл раствора соли Мора.
Титр раствора соли Мора определяют перед началом анализа (контрольное титрование).
Из приведенной выше реакции (1) следует, что 1 мл раствора двухромовокислого калия с содержанием 0,0426087 г соответствует 0,0126 мл спирта.
Содержание спирта в среде вычисляют по формуле
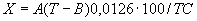 ,
,
где - содержание спирта, об. %; 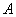 - количество двухромовокислого калия, взятое для анализа, мл; - количество соли Мора, пошедшее на титрование испытуемой бражки, мл; - количество соли Мора, пошедшее на контрольное титрование (титр соли Мора), мл; - объем бражки, взятый на определение, мл; 100 - коэффициент перевода, %; 0,0126 - коэффициент перевода KСrО на спирт, мл.
- количество двухромовокислого калия, взятое для анализа, мл; - количество соли Мора, пошедшее на титрование испытуемой бражки, мл; - количество соли Мора, пошедшее на контрольное титрование (титр соли Мора), мл; - объем бражки, взятый на определение, мл; 100 - коэффициент перевода, %; 0,0126 - коэффициент перевода KСrО на спирт, мл.
Пример. На определение взято 10 мл бражки и 25 мл KСrО. На контрольное титрование пошло 95 мл раствора соли Мора, а на титрование бражки - 90 мл раствора соли Мора. Определим содержание спирта (об. %)
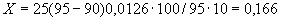 %.
%.
Ускоренный метод определения спирта (ПНР)
Быстрое определение спирта в культуральной среде позволяет оптимизировать процесс выращивания дрожжей.
Сущность метода, разработанного Э.Собчаком (ПНР), заключается в том, что горячий спирт пропускают через раствор двухромовокислого калия, подкисленного серной кислотой.
Избыток двухромовокислого калия после добавления йодистого калия определяют титрованием выделенного йода 1 н. раствором гипосульфита. Индикатором служит 0,4%-ный раствор крахмала.
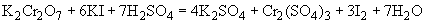 ;
;
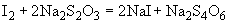 .
.
По расходу раствора гипосульфита при помощи табл.3 (см. приложение IV) определяют содержание спирта.
Ход определения заключается в следующем. В колбу 1
(рис.26) наливают 20 мл дистиллированной воды, а в приемник пипеткой вносят 10 мл раствора KСrO (42,59 г на 1 л) и 10 мл раствора HSO (относительной плотностью 1,84), разбавленные 1:1. Затем колбу 1
соединяют с приемником 2
стеклянной трубкой так, чтобы конец погруженной трубки находился почти над самым дном приемника с двухромовокислым калием. Затем колбу с водой медленно подогревают до кипения. Пар проходит через стеклянную трубку и конденсируется. Конденсат стекает в раствор двухромовокислого калия и нагревает его (наружу выходят пузырьки воздуха и водяного пара). Началом отгонки спирта считается момент закипания раствора двухромовокислого калия в приемнике, она продолжается 1-3 мин. Затем кипение прерывают, вынимают пробку из колбы для отгонки так, чтобы двухромовокислый калий, находящийся в приемнике, не был втянут в колбу. Содержимое приемника переносят количественно в коническую колбу с притертой пробкой, многократно ополаскивая небольшими порциями дистиллированной воды пробирку и трубку перегонной установки. Общий объем двухромовокислого калия вместе с водой, израсходованной на перенос раствора, должен составлять 50-70 мл.
Рис.26. Прибор для определения спирта ускоренным методом (ПНР)
Затем в коническую колбу добавляют 5 мл 10%-ного раствора йодистого калия, закрывают притертой пробкой и выдерживают в течение 2 мин. После этого содержимое колбы охлаждают до комнатной температуры и добавляют 2 мл 0,4%-ного раствора крахмала. Хорошо перемешанную пробу титруют 1 н. раствором гипосульфита (248,2 г NaSO·5НО на 1 л).
При проведении таким образом глухого опыта (с двумя повторностями) на титрование выделенного йода обычно расходуется 8-9 мл раствора гипосульфита. Затем определяют количество спирта. Если концентрация его выше 1%, то раствор разбавляют водой. При выращивании хлебопекарных дрожжей содержание этилового спирта в культуральной среде определяют (с двумя повторностями) так же, как и при проведении глухого опыта, с той лишь разницей, что в перегонную колбу вносят 10 мл исследуемого раствора и 10 мл дистиллированной воды. По разнице гипосульфита, израсходованного на титрование йода в глухом опыте () и в исследуемом образце (), по табл.3 (см. приложение IV) находят содержание в нем спирта.
Пример. При проведении глухого опыта израсходовано 8,3 мл гипосульфита, а при определении спирта в исследуемой пробе - 7,6 мл. Разница в расходе гипосульфита составит 8,3-7,6=0,7 мл. Этой разнице и расходу гипосульфита в глухом опыте (8,3 мл) в табл.3 (см. приложение IV) соответствует 0,11% спирта, содержащегося в культуральной среде.
Определение содержания растворенного в среде кислорода
В дрожжевой клетке ферментативные процессы могут быть направлены либо на образование спирта, либо на накопление биомассы, что обусловлено количеством растворенного кислорода в среде. При недостатке кислорода в среде одновременно протекают оба процесса. Экономический коэффициент использования сырья при этом понижается и тем сильнее, чем меньше кислорода получают дрожжевые клетки.
Потребность дрожжевых клеток в кислороде зависит от ряда факторов, которые изучаются в настоящее время. Известно только, что потребность в кислороде зависит от энергетических материалов, возраста клеток, условий формирования их дыхательной системы. И.П.Астаховой показано, что для образования 1 г молодых растущих клеток требуется 80-100 г кислорода в час, а более старых - 40-60 мг О/ч. По данным К.П.Андреева, потребность дрожжей в кислороде составляет 0,76-0,78 кг/кг. Известно также, что понижение концентрации кислорода в среде до 1 мг/л почти прекращает размножение дрожжевых клеток. По данным отечественных и зарубежных исследователей, активный процесс роста и размножения дрожжевых клеток наблюдается при концентрации кислорода в среде 2 мг/л.
Для определения растворенного в среде кислорода чаще применяют видоизмененный метод Винклера. Метод основан на ряде химических реакций. Сначала протекает реакция между NaOH и MnCl с образованием осадка гидрата закиси марганца.
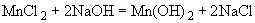 .
.
Затем, если в исследуемой жидкости содержится кислород, часть гидрата закиси марганца окисляется в гидрат окиси.
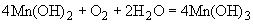 .
.
В среде образуется смесь гидратов закиси и окиси марганца, которые, реагируя с соляной или серной кислотой, образуют двух- и треххлористый марганец:
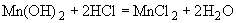 ;
;
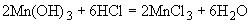 .
.
Треххлористый марганец в присутствии кислорода легко выделяет Сl, который разлагает йодистый калий.
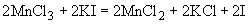 .
.
Выделившийся йод оттитровывают 0,01 н. раствором серноватисто-кислого натрия, по которому определяют количество растворенного в среде кислорода.
При содержании в воде большого количества органических веществ возможно связывание ими йода, вследствие чего получаются искаженные результаты, вплоть до отрицательных. В связи с этим устанавливают величину поправки на связывание йода веществами среды. Для этого ставят контрольную пробу.
Ход определения заключается в следующем. Пробы исследуемой жидкости для определения кислорода берут в две склянки вместимостью 150-300 мл с хорошо пришлифованными стеклянными пробками. Вместимость склянок точно определяют взвешиванием или предварительным калиброванием. Для каждого определения берут две склянки равного объема и наливают в них по 15 мл 0,1 н. раствора йода и по 3 мл раствора NaOH с KI. Затем в одну из склянок (контрольную) осторожно по стенке наливают исследуемую жидкость и закрывают, не оставляя ни одного пузырька воздуха; во вторую склянку наливают исследуемую жидкость и 2 мл раствора МnСl и склянку осторожно закрывают так, чтобы под пробкой не образовались пузырьки воздуха. Закрытую склянку хорошо перемешивают, переворачивая ее, до образования хлопьевидного осадка. Образовавшемуся осадку гидрата окиси и закиси марганца дают осесть как в опытной, так и контрольной склянке, в течение 30 мин. Затем склянки открывают, прозрачный раствор над осадком сливают с помощью сифона в конические колбы вместимостью по 500 мл. В склянки наливают по 5 мл разбавленной соляной или серной кислоты (2:3) и хорошо взбалтывают до полного растворения осадка. Раствор из склянок сливают в колбы; склянки ополаскивают дистиллированной водой, которую присоединяют к содержимому конических колб.
Весь раствор основного и контрольного опытов в присутствии крахмала титруют серноватистокислым натрием до исчезновения синей окраски; 1 мл 0,01 н. раствора гипосульфита соответствует 0,08 мг кислорода.
Количество кислорода , содержащееся в 1 л исследуемой жидкости в массовых единицах, рассчитывают по формуле
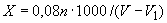 ,
,
где  ;
; 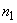 - количество 0,01 н. раствора NаSO, пошедшее на титрование в основном опыте, мл;
- количество 0,01 н. раствора NаSO, пошедшее на титрование в основном опыте, мл; 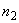 - количество 0,01 н. раствора NaSО, пошедшее на титрование в контрольной пробе, мл; - вместимость склянки, мл; - количество реактивов, введенное в склянку, мл.
- количество 0,01 н. раствора NaSО, пошедшее на титрование в контрольной пробе, мл; - вместимость склянки, мл; - количество реактивов, введенное в склянку, мл.
Определение скорости роста дрожжей
Скорость размножения дрожжей при их выращивании на мелассовых средах может колебаться в широких пределах (от 0,05 до 0,371), что зависит как от физиологического состояния клеток, так и условий их культивирования.
Интенсивность накопления биомассы характеризуется скоростью роста и скоростью размножения. Скорость роста указывает на увеличение массы клеток, а скорость размножения - частоту их почкования. Для оценки процесса роста часто пользуются средней скоростью роста 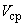 , под которой понимают количество биомассы, накопившей за определенный промежуток времени. Ее вычисляют по формуле
, под которой понимают количество биомассы, накопившей за определенный промежуток времени. Ее вычисляют по формуле
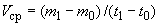 ,
,
где - количество дрожжей в начале процесса; - в конце промежутка времени 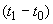 .
.
Дрожжи - это биологический материал, в котором клетки различаются как по возрасту и величине, так и по физиологическому состоянию. В этой связи обычно пользуются понятием "относительная" (удельная) скорость роста 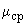 , определяемая по формуле
, определяемая по формуле
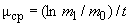 ,
,
где - количество дрожжей в начале процесса, кг; - количество дрожжей, полученных за время , кг.
Пример. Для воздушно-проточного способа культивирования дрожжей
I. Накопительная стадия
- 1100 кг - количество дрожжей, засеянных в дрожжерастильный аппарат;
- 3400 кг - получено дрожжей в конце накопительного периода;
- 7 ч - длительность накопительного периода.
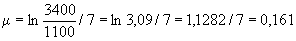 .
.
Логарифм 3,09=1,1282, его находим по таблице натуральных логарифмов.
II. Стадия непрерывного роста дрожжей
- 4000 кг - получено дрожжей в конце определенного часа наблюдения;
- 3400 кг - было дрожжей в начале этого часа;
- 1 ч.
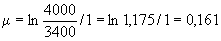 .
.
Для характеристики процесса роста и размножения дрожжей часто пользуются коэффициентом часового прироста биомассы , который показывает, во сколько раз биомасса увеличилась в течение 1 ч. Если обозначить количество биомассы в начале процесса через количество биомассы, полученное через 1 ч , через 2 ч , через 3 ч 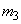 и т.д., то коэффициент часового прироста биомассы выразится следующим образом:
и т.д., то коэффициент часового прироста биомассы выразится следующим образом:
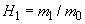 ;
; 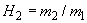 ;
; 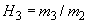 и т.д.
и т.д.
Пример. В дрожжерастильный аппарат засеяли 1000 кг дрожжей. Через час количество биомассы составило 1160 кг. При этом коэффициент часового прироста дрожжей составит =1160/1000=1,16, а удельная скорость роста 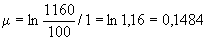 .
.
 находим по таблице натуральных логарифмов.
находим по таблице натуральных логарифмов.
Определение содержания нитритов
Нитритообразующие бактерии приносят большой вред размножению дрожжевых клеток. В процессе своей жизнедеятельности они восстанавливают нитраты (соли азотной кислоты) в нитриты (соли азотистой кислоты), которые являются ядом для дрожжевых клеток. По данным Е.А.Плевако, при концентрации нитритов 0,0005% наблюдается задержка роста дрожжей, а при концентрации более 0,004% клетки отмирают, причем в первую очередь погибают почки и молодые, только отпочковавшиеся клетки.
Определение содержания нитритов в культуральной среде и дрожжевом молоке проводят по методике, разработанной в ХО ВНИИХП. Предварительно наличие нитритов устанавливают качественной пробой с реактивом Грисса.
Количественное определение нитритов с помощью фотоэлектроколориметра (ХО ВНИИХП). При оценке содержания нитритов в среде в процессе размножения дрожжей на разных стадиях их производства, в среде или дрожжевом молоке "много"-"очень много", в мерную колбу вместимостью 100 мл отбирают 1-4 мл испытуемого раствора, добавляют 0,5-1 г активного угля, доводят до метки дистиллированной водой, хорошо перемешивают, выдерживают 5-10 мин и фильтруют через плотный фильтр (если среда не очень темная, осветление активированным углем необязательно).
40 мл фильтрата отбирают в мерную колбу вместимостью 50 мл, добавляют 8 мл реактива Грисса, доводят до метки водой, взбалтывают и спустя 15 мин определяют на фотоэлектроколориметре оптическую плотность окрашенного в розовый цвет раствора при зеленом светофильтре в кюветах с расстоянием между гранями 5 мм.
Одновременно в двух колбах вместимостью 50 мл готовят стандартные растворы с различным содержанием NO (10-20 мл 2-го стандартного раствора NaNO) и также устанавливают оптическую плотность обоих растворов.
В случае оценки содержания нитритов в среде при размножении дрожжей или в бражке "мало"-"очень мало" в мерную колбу вместимостью 100 мл отбирают 10-20 мл испытуемого раствора и определение ведут так же, как описано при содержании нитритов "много"-"очень много".
Количество нитритов рассчитывают по формуле
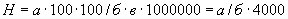 ,
,
где - содержание NO в 100 мл испытуемого раствора, %; - количество NO, соответствующее найденной оптической плотности раствора, мкг; - количество фильтрата, взятое для определения NO, мл; - количество испытуемого раствора, взятое на определение, мл; 100 - коэффициент пересчета на 100 мл осветленной среды и на 100 мл испытуемого раствора; 1000000 - коэффициент пересчета микрограммов в граммы.
Пример. Качественная проба на нитриты оценена "много" и на определение взято 3 мл испытуемого раствора в мерную колбу вместимостью 100 мл. После фильтрации 40 мл раствора отобрано в мерную колбу вместимостью 50 мл. Найденная оптическая плотность раствора составляет 0,134, оптическая плотность стандартного раствора, содержащего 30 мкг, равна 0,159. Определяем количество NO (в мкг), соответствующее найденной оптической плотности,
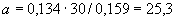 мкг NO.
мкг NO.
Содержание NO в 100 мл взятой среды или бражки составит
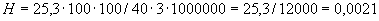 % NO.
% NO.
Количественное определение нитритов в среде или дрожжевом молоке визуальным способом (ХО ВНИИХП). При визуальном способе определения содержания нитритов в среде на различных стадиях производства или дрожжевом молоке также предварительно устанавливают наличие нитритов качественной пробой с реактивом Грисса.
При оценке содержания нитритов "много"-"очень много" в мерную колбу вместимостью 100 мл отбирают 1-4 мл среды, добавляют 0,5-1,0 г активного угля, доводят до метки дистиллированной водой, перемешивают, выдерживают 5-10 мин и фильтруют через плотный фильтр. (Если среда не очень темная, осветление активным углем необязательно.)
После этого в 4 пробирки одинакового диаметра и объема отбирают 1, 2, 3, 4 мл прозрачного фильтрата, добавляют соответственно 3,3, 2,2, 1,2, 0,2 мл дистиллированной воды до объема 4,2 мл. Затем в каждую из пробирок добавляют по 0,8 мл раствора Грисса и тщательно взбалтывают.
Одновременно готовят 4 пробирки со стандартным раствором NaNO, отбирая в каждую пробирку 1, 2, 3, 4 мл стандартного раствора. В каждой пробирке таким образом находится 1, 2, 3, 4 мкг NО (пробирки с большим содержанием NO готовить не следует). В пробирки соответственно добавляют 3,2; 2,2; 1,2; 0,2 мл дистиллированной воды и по 0,8 мл в каждую пробирку раствора Грисса и хорошо перемешивают. Спустя 15 мин устанавливают, в какой из 4 пробирок со стандартным раствором совпадает розовая окраска испытуемого раствора.
Для большой точности определения количества нитритов можно взять не 4 пробирки со стандартным раствором, а 8 и соответственно набирать в каждую 0,5; 1; 1,5; 2; 2,5; 3; 3,5; 4 мл стандартного раствора, в которых находится от 0,5 до 4 мкг NO.
При оценке качественной реакции на нитриты "мало"-"очень мало" отбирают 10-20 мл испытуемого раствора и определение ведут так же, как описано при содержании нитритов "много"-"очень много".
Содержание нитритов рассчитывают по формуле, приведенной выше, в которой обозначает количество NO, найденное в пробирке (в мкг).
Пример. На анализ взято 2 мл испытуемого раствора (т.е. =2). После фильтрации на определение в пробирки отобрано по 4 мл (т.е. =4).
Окраска раствора в пробирках совпала с окраской 5-й пробирки стандартного ряда, что соответствует 2,5 мкг NO (т.е. =2,5). Определяем содержание нитритов в 100 мл испытуемого раствора.
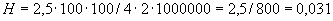 % NO.
% NO.
КОНТРОЛЬ ПРОЦЕССА ВЫДЕЛЕНИЯ ДРОЖЖЕЙ
По окончании процесса выращивания дрожжи выделяют из культуральной среды. Сначала их отделяют от бражки на сепараторах, затем несколько раз промывают холодной водой для удаления продуктов метаболизма, сгущают и получают дрожжевое молоко с содержанием 300-700 г/л дрожжей. Окончательное отделение дрожжей происходит на вакуум-фильтрах или фильтр-прессах.
Контроль осуществляется за чистотой промывки дрожжей. Учитывают потери дрожжей при сепарировании и прессовании, а также степень отмывки дрожжей от продуктов метаболизма (окисляемость).
Определение потерь дрожжей
Потери дрожжей определяют визуально или путем микроскопирования фильтрата, отходящего от сепараторов и вакуум-фильтров.
Визуальные методы. 1. Капля фильтрата, стекающая с пальцев, при просмотре на свет должна быть совершенно прозрачной. 2. Фильтрат, налитый в пробирку, после отстаивания в течение 2-3 мин должен быть совершенно прозрачным.
Микроскопирование. Пробу, отходящую от сепараторов и вакуум-фильтров, просматривают под микроскопом (не менее 5 полей зрения). При хорошей работе сепараторов и вакуум-фильтров в фильтрате наблюдается 1-2 клетки дрожжей не в каждом поле зрения. Допускается не более 5-7 клеток не в каждом поле зрения.
Определение чистоты промывки дрожжей
Степень промывки дрожжей определяют по окисляемости промывной воды, которую определяют либо титрованием, либо специальным прибором, разработанным работниками Курганского дрожжевого завода О.И.Феофоновым и А.А.Прудниковым.
Метод титрования.
Для определения берут 10 мл отфильтрованной от дрожжей жидкости, добавляют 0,5 мл раствора HSO (1:3) и титруют 0,2%-ным раствором KMnO до слабо-розового окрашивания. Величина окисляемости выражается количеством KMnO, пошедшим на титрование. У хорошо промытых дрожжей окисляемость составляет 0,1-0,15 мл раствора KMnO. Предельно допустимая норма окисляемости 0,25 мл.
Быстрый метод определения окисляемости специальным прибором. Действие прибора основано на измерении силы тока, проходящего через промывную воду.
Принципиальная схема прибора представлена на рис.27. Промывную воду наливают в стакан с крышкой из изоляционного материала. Крышку с вмонтированными в нее двумя электродами вставляют в стакан и прибор включают в сеть. Через жидкость проходит электрический ток, его величина регистрируется на шкале прибора, которая градуирована в единицах прибора, показывающих степень промывки дрожжей (шкала градуируется опытным путем). Показания прибора не зависят от температуры промывной жидкости и уровня жидкости, взятой для определения. Прибор удобен в эксплуатации и позволяет быстро определить степень промывки дрожжей.
Рис.27. Принципиальная схема прибора для определения степени промывки дрожжей
АНАЛИЗ ПРЕССОВАННЫХ ДРОЖЖЕЙ
Определение влажности
Арбитражный метод. Пробу дрожжей (не менее 10 г) протирают через сетку с отверстиями диаметром 2-3 мм или крошат ножом, взвешивают две навески по 1,5 г на аналитических весах в чистой, предварительно высушенной и доведенной до постоянной массы бюксе с притертой крышкой и сушат в сушильном шкафу при температуре 105 °С до постоянной массы. Первый раз дрожжи взвешивают через 4 ч, затем через 1 ч после высушивания. Дрожжи сушат до тех пор, пока разница в массе между двумя взвешиваниями не превысит 0,001 г (при вычислении результатов доли до 0,05% отбрасывают, а доли, равные 0,05 и более, округляют до 0,1).
Навеску высушивают в открытой бюксе (крышку помещают рядом с бюксой). Прежде чем вынуть бюксу из сушильного шкафа, ее закрывают крышкой, а затем охлаждают в эксикаторе в течение 20-30 мин. Так поступают перед каждым взвешиванием.
Влажность в процентах (в %) вычисляют по формуле
 ,
,
где - масса дрожжей до высушивания, г; - масса дрожжей после высушивания, г.
Ускоренный метод (ХО ВНИИХП). Для внутризаводского контроля рекомендуется ускоренный метод определения влажности прессованных дрожжей.
В доведенной до постоянной массы бюксе на аналитических весах взвешивают 2,5-3,5 г измельченных дрожжей (протертых через сетку с диаметром 2-3 мм) и высушивают в течение 70 мин в сушильном шкафу при температуре 130 °С. После этого бюксы вынимают, накрывают крышками, охлаждают в эксикаторе и взвешивают на аналитических весах.
Влажность (в %) вычисляют по формуле, приведенной выше.
Пример. Масса бюксы с дрожжами до высушивания 32,0929 г, масса пустой бюксы 29,4998 г, масса бюксы с дрожжами после высушивания 30,1541 г,
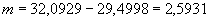 г;
г;
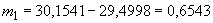 г;
г;
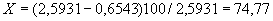 %.
%.
Метод точен, дает результаты, хорошо совпадающие с определением влажности дрожжей арбитражным методом. Отклонение при определении колеблется в пределах ±(0,24-0,35)%, что вполне отвечает требуемой точности анализа.
Предварительная подсушка прессованных дрожжей при 30 °С не рекомендуется, так как приводит к потере 2-3% сухого вещества дрожжей.
Рекомендуемый метод позволяет одновременно определить влажность нескольких партий дрожжей.
Определение подъемной силы дрожжей
Подъемная сила является одним из основных показателей качества дрожжей. Чем быстрее дрожжи поднимают тесто, тем лучше их качество. Хорошие дрожжи поднимают тесто за 45-60 мин. В соответствии с требованиями стандарта подъемная сила товарных дрожжей не должна превышать 70 мин. На величину подъемной силы дрожжей могут оказывать небольшое влияние влажность и качество муки.
Ход определения заключается в следующем. 280 г пшеничной муки 85%-ного помола (ОСТ КЗ СНК 8470/268) нагревают в термостате при 35 °С в течение 2 ч. Нагревают до 35 °С 160 мл 2,5%-ного раствора поваренной соли (раствор готовят на водопроводной воде). На технохимических весах (с точностью до 0,01 г) отвешивают 5 г анализируемых прессованных дрожжей. В фарфоровой чашке (35 °С) смешивают 5 г дрожжей с 15-20 мл подогретого до 35 °С раствора поваренной соли до получения однородной суспензии. Разведенные дрожжи быстро выливают в дежу лабораторной тестомесильной машины, замешивающей тесто при частоте вращения 135 об/мин. Оставшимся раствором соли споласкивают чашку из-под дрожжей и выливают его в дежу тестомесильной машины. Затем насыпают 280 г муки, подогретой до 35 °С, и замешивают тесто в течение 5 мин. Тесту придают форму батона (по размеру формочки) и тотчас же переносят его в специальную металлическую форму, предварительно нагретую в термостате при температуре 35 °С и смазанную растительным маслом. Форма должна представлять собой в продольном и поперечном разрезах трапеции следующих внутренних размеров: верхние основания 14,3 и 9,2 см, нижние - 12,6 и 8,5 см, высота 8,5 см.
После перенесения теста в форму на длинные борта ее навешивают поперечную металлическую перекладину, входящую в форму на 1,5 см. Форму помещают в термостат с температурой 35 °С.
Подъемной силой считают время (в мин), в течение которого тесто, помещенное в форму, соприкоснется с нижним краем перекладины, т.е. подъемом теста до 70 мм.
При отсутствии тестомесильной машины тесто замешивают вручную, соблюдая указанный выше режим приготовления его.
Определение кислотности
10 г прессованных дрожжей отвешивают на технохимических весах. В фарфоровой чашке или стакане готовят суспензию с 50 мл дистиллированной воды и титруют 0,1 н. раствором NaOH в присутствии индикатора фенолфталеина до появления розового окрашивания, не исчезающего в течение нескольких секунд.
Кислотность дрожжей (в мг уксусной кислоты) рассчитывают по формуле
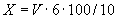 ,
,
где - количество 0,1 н. раствора щелочи, пошедшее на нейтрализацию, мл; 6 - количество мг уксусной кислоты, соответствующее 1 мл 0,1 н. раствора щелочи.
Примечание. При вычислении результатов анализа доли до 0,5 отбрасывают, а доли, равные 0,5 и выше, округляют до единицы.
Кислотность хороших дрожжей обычно находится в пределах 60-120 мг уксусной кислоты на 100 г прессованных дрожжей.
Пример. На титрование 10 г прессованных дрожжей затрачено 2,0 мл 0,1 н. раствора гидроксида натрия, что соответствует 2,0·6·100/10=120 мг уксусной кислоты на 100 г дрожжей.
Определение стойкости дрожжей
По этому показателю определяют сохраняемость прессованной продукции.
Определение стойкости дрожжей при хранении в холодильнике.
От каждой партии дрожжей одной даты выработки в этот день отбирают бруски дрожжей, укладывают в применяемую для отправки тару и помещают в холодильную камеру с температурой 0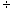 +4 °С. На этикетке бруска дрожжей отмечают час и дату начала хранения. За пробой ведут наблюдение 10 сут, в течение этого срока ежедневно отмечают в журнале органолептические показатели дрожжей. Через 10 сут хранения определяют кислотность дрожжей и результаты анализа записывают в лабораторном журнале. Согласно требованиям ГОСТа на дрожжи они должны выдержать хранение в течение 10 сут.
+4 °С. На этикетке бруска дрожжей отмечают час и дату начала хранения. За пробой ведут наблюдение 10 сут, в течение этого срока ежедневно отмечают в журнале органолептические показатели дрожжей. Через 10 сут хранения определяют кислотность дрожжей и результаты анализа записывают в лабораторном журнале. Согласно требованиям ГОСТа на дрожжи они должны выдержать хранение в течение 10 сут.
Определение стойкости дрожжей при хранении в термостате (ускоренный метод). Завернутый в бумагу брусок дрожжей массой 0,5 или 1 кг, предварительно охлажденный до +4 °С, помещают в негерметизированный термостат при 35 °С и хранят до полного размягчения. Время в часах, прошедшее от момента помещения дрожжей в термостат до их размягчения, характеризует стойкость дрожжей.
Стойкие при хранении дрожжи в этих условиях сохраняются более 100 ч и высыхают. Иногда дрожжи размягчаются через 40 ч и ранее, такие дрожжи считаются непригодными для дальнейших перевозок. Согласно ГОСТ 171-81 стойкость дрожжей должна быть не менее 60 ч.
Определение стойкости дрожжей по изменению их подъемной силы (ускоренный метод). О стойкости дрожжей также можно судить по изменению подъемной силы дрожжей, определяемой одновременно в дрожжах, хранившихся в термостате и холодильнике.
Ход определения заключается в следующем. Берут две навески дрожжей по 5 г и помещают в маленькие тигли, один ставят в термостат на 24 ч при 35 °С (требуется точно соблюдать температуру и продолжительность хранения), другой - в холодильную камеру (контроль). По истечении этого срока дрожжи вынимают из тиглей и определяют подъемную силу. Стойкость определяют по разности подъемной силы дрожжей, хранившихся в термостате и холодильнике. Стойкость дрожжей считается хорошей, если подъемная сила ухудшается не более 20 мин.
Определение содержания общего азота в дрожжах
Дрожжи содержат различное количество азота, что характеризуется стадией их культивирования. Так, при получении дрожжей чистой культуры на первых двух стадиях в дрожжах содержится 2,8% азота, на третьей - 2,6%, на четвертой - 2,5%. Дрожжи ЕЧК в первой стадии обычно содержат 2,4% азота, во второй - 2%. Товарные дрожжи на первой стадии (Б) содержат 1,9% азота, а на второй стадии (В) - 1,6-1,8%. Причем дрожжи, предназначенные для сушки, обычно содержат не более 1,6% азота, а предназначенные для упаковки - 1,8%.
Анализируют каждую партию дрожжей. Общий азот в прессованных дрожжах определяют по методу Кьельдаля.
Ход определения. Около 4 г прессованных дрожжей отвешивают в бюксе на аналитических весах с точностью до четвертого знака и переносят в колбу Кьельдаля. Остаток дрожжей в бюксе взвешивают и по разности определяют массу взятой навески. В колбу наливают 20 мл концентрированной, химически чистой серной кислоты, добавляют 2-3 крупинки селена или около 1 г сернокислой меди и около 10 г сернокислого калия (катализаторы). Если в дальнейшем в этой же навеске дрожжей определяют и фосфор, сжигание с медью производить нельзя.
Сжигание дрожжей и отгонку азота проводят так же, как и при определении общего азота в мелассе. Если параллельно с определением азота определяют фосфор, то из мерной колбы сначала отбирают 50 мл для этого определения, а остальной раствор переводят в колбу для отгона аммиака.
Содержание азота (в %) вычисляют по формуле
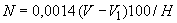 ,
,
где - количество 0,1 н. раствора серной кислоты, налитое в приемник, мл; - количество 0,1 н. раствора щелочи, затраченное на титрование, мл; - навеска дрожжей, г; 0,0014 - коэффициент перевода миллилитров серной кислоты в граммы азота (1 мл 0,1 н. раствора серной кислоты соответствует 0,0014 г азота).
Общее количество белка вычисляют путем умножения количества общего азота на коэффициент 6,25.
Пример. Для анализа взято 4,8200 г дрожжей. Для определения фосфора отобрана 1/5 часть, что составляет 4,8200/5=0,964 г. Для определения азота взято 4,8200-0,964=3,8560 г. В приемник налито 75 мл 0,1 н. раствора серной кислоты. На титрование оставшейся свободной кислоты затрачено 22,4 мл 0,1 н. раствора гидроксида натрия. Аммиаком связано 75-22,4=52,6 мл кислоты. Следовательно, содержание азота в дрожжах составит
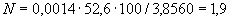 %.
%.
Определение содержания фосфора
Содержание фосфора в дрожжах, как и азота, зависит от стадии их культивирования. В первых трех стадиях при получении чистой культуры в дрожжах содержится 1,4% РО, в четвертой - 1,3% РО. В дрожжах ЕЧК на первой стадии содержится 1,2% РО, а на второй - 1,0% РО. Товарные дрожжи на стадии Б содержат 0,9% РО, а на стадии В - 0,8% РО. Фосфорный ангидрид в дрожжах определяют колориметрическим методом Бригса или при помощи фотоэлектроколориметра.
Метод Бригса.
Принцип метода основан на образовании фосфорномолибденовой кислоты и ее восстановлении гидрохинином и NaSO с образованием устойчивой синей окраски. Интенсивность окраски возрастает пропорционально количеству неорганического фосфора в испытуемой пробе.
Ход определения. Навеску прессованных дрожжей около 1 г помещают в колбу Кьельдаля и сжигают так же, как это указано при определении общего азота. После сжигания содержимое колбы переводят в мерную колбу вместимостью 100 мл. Колбу Кьельдаля несколько раз ополаскивают водой, которую также сливают в мерную колбу во избежание потерь. Затем содержимое колбы доводят дистиллированной водой приблизительно до половины объема мерной колбы, нейтрализуют 33%-ным раствором гидроксида натрия, охлаждают, доливают дистиллированной водой до метки и хорошо перемешивают. Из мерной колбы отбирают 1 мл в пробирку высотой 70 мм и диаметром 15 мм.
Одновременно в семи других пробирках такого же размера готовят стандартный ряд со все увеличивающимися концентрациями фосфора: 0,01; 0,02, 0,03; 0,04; 0,05; 0,06; 0,07 мг фосфора в 1 мл. Для этого в первую из семи пробирок наливают 0,1 мл стандартного раствора KHPO, что соответствует 0,01 мг фосфора: во вторую - 0,2 мг, т.е. 0,02 мг фосфора; в третью - 0,3; в четвертую - 0,4, в пятую - 0,5; в шестую - 0,6; седьмую - 0,7 мл. Затем в пробирки наливают дистиллированную воду: в первую 0,9 мл, во вторую 0,8, в третью 0,7, в четвертую 0,6, в пятую 0,5, в шестую 0,4, в седьмую 0,3 мл. Таким образом, в каждой из семи пробирок объем жидкости составляет 1 мл.
Затем в каждую из восьми пробирок (семь стандартного ряда и одна с испытуемым раствором) наливают по 1 мл молибденового раствора, по 1 мл раствора гидрохинона и по 1 мл раствора безводного сульфита и хорошо взбалтывают, после чего содержимое пробирок окрашивается в синий цвет различной интенсивности - от светло-синего до темно-синего. Пробирки оставляют в покое на 20 мин, после чего быстро сравнивают окраску пробирки с испытуемой жидкостью с окраской каждой из семи пробирок стандартного раствора.
Содержимое фосфора (в %) рассчитывают по формуле
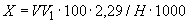 ,
,
где - содержание фосфора в пробирке с испытуемым веществом (по стандартному ряду), мг; - разведение; - масса дрожжей в пересчете на 25% СВ, г; 2,29 - коэффициент перевода в РО (частное от деления молекулярной массы РО на молекулярную массу фосфора 142/62=2,29); 100 - пересчет на проценты; 1000 - перевод в граммы.
Пример. На определение взят 1 г прессованных дрожжей с содержанием 25% СВ. После сжигания в колбе Кьельдаля все содержимое перенесено в мерную колбу на 100 мл. На определение фосфора взят 1 мл раствора. После сравнения окраски испытуемого раствора с цветом растворов стандартного ряда установили, что цвет пробирки с испытуемым раствором соответствует цвету пробирки стандартного ряда, содержащей 0,04 мг фосфора.
Содержание PO в прессованных дрожжах составит
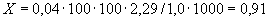 %.
%.
Уточненный метод Фиске и Сабуро (ХО ВНИИХП). Метод Фиске и Сабуро широко распространен в аналитической практике. Для упрощения этого метода введен ряд изменений. В качестве восстановителя вместо эйконогена принята аскорбиновая кислота, устранен подогрев при 37 °С, общий объем всех растворов составляет 5 мл, а не 50. Интенсивность окраски устанавливают визуально или на фотоэлектроколориметре.
При визуальном способе определение интенсивности синей окраски устанавливают сопоставлением окраски испытуемого раствора с окраской ряда пробирок стандартного раствора однозамещенного фосфорнокислого калия (KНРО) с содержанием от 0,01 до 0,08 мг фосфора.
При определении интенсивности окрашивания растворов на фотоэлектроколориметре сначала строят калибровочный график. Для этого готовят пробирки стандартного ряда и каждую из них колориметрируют, определяя оптическую плотность раствора. На оси абсцисс откладывают количество фосфора в миллиграммах, на оси ординат - соответствующую ему оптическую плотность.
Содержание фосфора в дрожжах выражают в пересчете на фосфорный ангидрид РО (в %).
Подготовку дрожжей и ход определения проводят следующим образом. В прессованных или сушеных дрожжах содержание общего фосфора определяют после минерализации дрожжей, т.е. в виде неорганического фосфора. Для этого дрожжи измельчают на сите, отвешивают в стаканчик на аналитических весах около 2 г, переносят их в колбу Кьельдаля вместимостью 100 мл, добавляют 8 мл концентрированной серной кислоты и озоляют. Катализатором при озолении служит 30%-ный пергидроль. Его осторожно несколько раз добавляют по 2-3 мл, предварительно охладив колбы Кьельдаля.
После полного обесцвечивания раствора колбу Кьельдаля смывают 5-8 мл воды и продолжают нагрев до появления белых паров (для полного удаления перекиси водорода). Затем колбу Кьельдаля охлаждают и раствор количественно переводят в мерную колбу вместимостью 100 мл, охлаждают до 20° С, доводят до метки и перемешивают.
Для дальнейшего определения фосфора требуется тщательная нейтрализация раствора. Так как добавка индикатора при нейтрализации может в дальнейшем влиять на оттенок синей окраски, в отдельной порции устанавливают количество щелочи, расходуемой на нейтрализацию. Для этого пипеткой отбирают в колбу 5 мл раствора и оттитровывают его по метиловому красному 10%-ным раствором NaOH.
Затем в мерную колбу на 50 мл отбирают пипеткой 25 мл кислого раствора озоленных дрожжей и добавляют пятикратное количество щелочи, пошедшее на нейтрализацию 5 мл этого раствора. После этого колбу охлаждают до 20° С, доводят до метки водой и хорошо перемешивают.
При нейтрализации нужно следить за тем, чтобы не было излишка щелочи, так как это вызывает появление мути.
Определение на фотоэлектроколориметре. В пробирку отбирают 1 мл отнейтрализованного раствора озоленных дрожжей, добавляют 2 мл дистиллированной воды и 1 мл раствора молибденовокислого аммония, тщательно взбалтывают, добавляют 1 мл 0,2%-ного раствора аскорбиновой кислоты, опять взбалтывают и выдерживают 20 мин при 20 °С. Общий объем жидкости в пробирке составляет 5 мл. Спустя 20 мин определяют на ФЭК оптическую плотность раствора в кюветах с расстоянием между гранями 5 мм при красном светофильтре. В кювету сравнения наливают дистиллированную воду. По калибровочному графику и найденной оптической плотности находят по оси абсцисс количество фосфора в миллиграммах.
При отсутствии ФЭК содержание фосфора определяют визуально.
Визуальный способ.
Отбирают 8-10 пробирок одинакового диаметра и объема и готовят в них стандартный ряд с увеличивающимся количеством фосфора: от 0,01 до 0,06 мг. Для этого в первую пробирку из колбы с 50 мл стандартного раствора KНРО отбирают 0,1 мл, что соответствует 0,01 мг фосфора, во вторую - 0,2 мл, т.е. 0,02 мг фосфора, и т.д., в шестую - 0,6 мл, т.е. 0,06 мг фосфора.
В первую пробирку добавляют 0,9 мл дистиллированной воды и в последующие - соответственно от 0,8 до 0,4 мл. Таким образом, в каждой пробирке находится 1 мл. Затем в отдельную пробирку из мерной колбы с 50 мл испытуемого нейтрального раствора озоленных дрожжей отбирают 1 мл. В каждую пробирку со стандартным и испытуемым раствором добавляют 2 мл дистиллированной воды и по 1 мл молибдата аммония, взбалтывают, выдерживают 20 мин при 20° С, после чего устанавливают, с какой пробиркой стандартного ряда совпадает синяя окраска испытуемого раствора с дрожжами.
Для большей точности определения фосфора визуальным способом можно взять 10-12 пробирок и отобрать в первую 0,1 мл раствора KНРО, что соответствует 0,01 мг фосфора, во вторую - 0,15 мл, т.е. 0,015 мг фосфора, и т.д., т.е. в четвертую - 0,25 мл, или 0,025 мг фосфора, и в десятую - 0,55 мл, т.е. 0,055 мг фосфора.
Дистиллированной воды следует добавить в каждую пробирку до объема 1 мл, т.е. в первую 0,9 мл, во вторую 0,85, в четвертую 0,75 и в десятую 0,45 мл воды.
АНАЛИЗ ДРОЖЖЕВОГО МОЛОКА
Анализы проводят по показателям ОСТ 18-369-81 на молоко дрожжевое. Анализируют среднюю пробу от каждой партии. Среднюю пробу отбирают после тщательного перемешивания в количестве не менее 1 л.
Определение концентрации дрожжей в дрожжевом молоке
Арбитражный метод. Из средней пробы после тщательного перемешивания отбирают 200 мл дрожжевого молока и фильтруют через два бумажных фильтра на воронке Бюхнера. Фильтрацию продолжают до тех пор, пока консистенция пласта дрожжей на фильтре не будет примерно соответствовать консистенции формованных дрожжей. Отфильтрованные дрожжи взвешивают с погрешностью до 0,01 г вместе с фильтровальной бумагой (во избежание потерь). Нижний кружок фильтровальной бумаги при взвешивании кладут на чашку весов с гирями, чтобы внести поправку к массе отфильтрованных дрожжей. В отфильтрованных дрожжах определяют влажность арбитражным методом (высушивание до постоянной массы).
Концентрацию дрожжей (в г/л) в пересчете на 25% СВ вычисляют по формуле
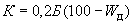 ,
,
где  - масса дрожжей, отфильтрованных из 200 мл дрожжевого молока, г;
- масса дрожжей, отфильтрованных из 200 мл дрожжевого молока, г;  - влажность отфильтрованных дрожжей, %.
- влажность отфильтрованных дрожжей, %.
Концентрация дрожжей в 1 л дрожжевого молока с влажностью 75% должна быть не менее 450 г.
Ускоренный метод. Концентрацию дрожжей в дрожжевом молоке определяют по содержанию в нем сухих веществ. Сухие вещества определяют сахарометром типа Е (0-8% СВ, 8-16% СВ и 16-24% СВ).
Дрожжевое молоко хорошо перемешивают мешалкой в течение 30 мин, отбирают среднюю пробу, доводят ее до температуры 20 °С, освобождают от пузырьков воздуха, если они имеются, и наливают в цилиндр до такого уровня, чтобы при погружении сахарометра часть ее выливалась из цилиндра в подставленную кювету. Часть дрожжевого молока, стекающая из цилиндра при погружении сахарометра, увлекает с собой пену, в результате чего ясно виден мениск. Наличие пены и пузырьков воздуха в дрожжевом молоке искажает результаты определения. Цилиндр устанавливают на строго горизонтальной поверхности. Диаметр цилиндра подбирают в соответствии с диаметром сахарометра, так как он не должен касаться стенок цилиндра.
Определив сахарометром плотность раствора, по табл.4 (см. приложение IV) находят концентрацию дрожжей (в г/л), соответствующую влажности прессованных дрожжей 75%.
Концентрация дрожжей в 1 л дрожжевого молока (в пересчете на прессованные дрожжи влажностью 75%) должна составлять не менее 450 г/л.
Определение количества дрожжей в дрожжевом молоке
Измеряют общий объем дрожжевого молока в сборнике, определяют концентрацию дрожжей в дрожжевом молоке методами, описанными выше. Количество дрожжей определяют по формуле
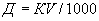 ,
,
где - количество прессованных дрожжей (25% СВ) в сборнике дрожжевого молока, кг; - концентрация дрожжей (25% СВ), определенная по табл.4 (см приложение IV), г/л; - объем дрожжевого молока в сборнике, м; 1000 - коэффициент перевода граммов в килограммы.
Определение подъемной силы
Для определения подъемной силы дрожжей можно брать либо отфильтрованный пласт дрожжей, либо тщательно перемешанное дрожжевое молоко.
Арбитражный метод. В отфильтрованном пласте дрожжей подъемную силу определяют по ГОСТ 171-81, а в дрожжевом молоке - следующим образом: 280 г пшеничной муки II сорта выдерживают в термостате при температуре 35 °С не менее 2 ч.
В фарфоровую чашку (35 °С) наливают дрожжевое молоко из средней пробы, количество которого зависит от концентрации дрожжей, определенной по табл.4 (см приложение IV), графы 3 и 8 (в пересчете на прессованные их должно быть 5 г). К дрожжевому молоку приливают 100 мл подогретой до 35 °С водопроводной воды и 4 г поваренной соли, перемешивают и снова добавляют воду в количестве, соответствующем разности между 160 мл и количеством дрожжевого молока, взятого на анализ. Затем к содержимому фарфоровой чашки добавляют муку (35 °С), замешивают тесто в течение 5 мин. Тесту придают форму батона и кладут в смазанную растительным маслом и нагретую до 35 °С форму. Размеры формы стандартные (см. выше). В форму помещают перекладину, находящуюся на высоте 7 см от дна. Форму с тестом ставят в термостат при температуре 35 °С и наблюдают за подъемом теста. Подъемной силой считается время в минутах, в течение которого тесто поднимается до перекладины (на высоту 7 см). Подъемная сила не должна быть более 70 мин.
Ускоренный метод (по всплыванию шарика теста). Подъемную силу дрожжей определяют либо в дрожжевом молоке, либо в отфильтрованном пласте дрожжей.
Определение подъемной силы дрожжей в отфильтрованном пласте. Среднюю пробу дрожжевого молока отфильтровывают на воронке Бюхнера, берут навеску дрожжей массой 0,31 г, помещают ее в предварительно нагретую до 35 °С фарфоровую чашку, добавляют 6,5-8,5 г муки II сорта (35 °С) и 4,8 мл 2,5%-ного раствора поваренной соли (35 °С), быстро замешивают тесто и придают ему форму шарика, который не должен прилипать к рукам. Шарик опускают в стакан с водой температурой 35 °С, ставят в термостат при температуре 35 °С и выдерживают там до тех пор, пока шарик не всплывет на поверхность воды. Замечают время от момента опускания шарика в воду до момента его всплытия. Для сравнения со стандартным методом это время следует умножить на коэффициент 3,5, полученный эмпирически.
Определение подъемной силы дрожжей в дрожжевом молоке. В мерную колбу вместимостью 50 мл наливают среднюю пробу дрожжевого молока, количество которого определяют по табл.4, графы 4 и 9 (см. приложение IV) в зависимости от содержания прессованных дрожжей. Затем в колбу добавляют 30 мл воды, подогретой до 35 °С, и 1,25 г поваренной соли, перемешивают, доливают водой до метки и опять тщательно перемешивают. 5 мл полученной суспензии смешивают с 6,5-8,5 г (в зависимости от влажности) муки II сорта (35 °С), быстро замешивают тесто, придают ему форму шарика, который не должен прилипать к рукам. Далее поступают так же, как описано выше.
Определение кислотности
Кислотность определяют либо в отфильтрованном пласте дрожжей, либо в дрожжевом молоке.
Определение кислотности в отфильтрованном пласте дрожжей. Кислотность определяют по ГОСТ 171-81.
Определение кислотности в дрожжевом молоке . В фарфоровую чашку или колбу вместимостью 250 мл отбирают количество дрожжевого молока, соответствующее 10 г прессованных дрожжей. Количество дрожжевого молока зависит от концентрации дрожжей в нем и определяется по табл.4, графы 5 и 10 (см. приложение IV). Затем добавляют дистиллированную воду в количестве, соответствующем разности между 50 мл и количеством взятого на определение дрожжевого молока, тщательно размешивают и титруют 0,1 н. раствором NaOH в присутствии индикатора фенолфталеина до появления розового окрашивания, не исчезающего в течение нескольких секунд.
Кислотность дрожжевого молока в пересчете на уксусную кислоту (в мг) на 100 г дрожжей вычисляют по формуле
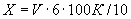 ,
,
где - количество 0,1 н. раствора NaOH, пошедшее на титрование, мл; 6 - количество уксусной кислоты, соответствующее 1 мл 0,1 н. раствора щелочи, мг; - поправочный коэффициент 0,1 н. раствора щелочи; 100 - переводной коэффициент, %; 10 - масса дрожжей, взятая на определение, г.
Кислотность выражают целым числом. Кислотность не должна быть более 120 мг уксусной кислоты на 100 г дрожжей в день выработки и не более 360 мг через 72 ч хранения при температуре от 0 до 10 °С.
КОНТРОЛЬ КАЧЕСТВА СУШЕНЫХ ДРОЖЖЕЙ
Определение влажности (арбитражный метод)
Около 2 г сушеных дрожжей взвешивают на аналитических весах в чистой, высушенной и тарированной бюксе с притертой крышкой. Бюксу с дрожжами переносят в сушильный шкаф, открывают крышку и сушат при температуре 105 °С до постоянной массы.
Содержание влаги (в %) вычисляют по формуле
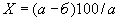 ,
,
где - масса навески до высушивания, г; - масса навески после высушивания, г.
Влажность сушеных дрожжей должна быть не выше 8%.
Примечание. Влажность сушеных дрожжей можно также определять экспресс-методами, например высушиванием в приборе "Влагомер В.И.Чижовой".
Определение подъемной силы (арбитражный метод)
К навеске сушеных дрожжей (2,5 г), взятой на технохимических весах, добавляют 30 мл водопроводной воды, нагретой до 43 °С, ставят в термостат при температуре 35 °С на 30 мин. После этого к размокшим дрожжам добавляют 15 г муки (пшеничной II сорта), тщательно размешивают и полученную смесь вновь ставят в термостат на 2 ч. Одновременно в термостат помещают 265 г муки с 130 мл воды, в которой растворено 4 г поваренной соли, и стандартного размера металлическую форму, смазанную растительным маслом.
Через 2 ч смесь с помощью 130 мл солевого раствора переводят в дежу лабораторной тестомесильной машины (или большую фарфоровую чашку), засыпают 265 г муки температурой 35 °С и замешивают тесто в течение 5 мин. Замешанное тесто вынимают, придают форму батона по размеру формочки и переносят в теплую металлическую форму (35 °С), на длинные бортики которой навешивают поперечную перекладину, входящую в форму на 1,5 см. Форму ставят в термостат (35 °С). Время (в мин), в течение которого тесто, помещенное в форму, соприкоснется с низким краем перекладины, называют подъемной силой дрожжей (размеры формы см. определение подъемной силы прессованных дрожжей). Подъемная сила хороших дрожжей I сорта должна быть не более 70 мин.
Определение пригодности сушеных дрожжей к длительному хранению
Опыт работы дрожжевой промышленности показывает, что о пригодности сушеных дрожжей к длительному хранению нельзя судить только по их подъемной силе и влажности. Для этой цели в сушеных дрожжах определяют содержание трегалозы, количество которой должно составлять 15-16% в расчете на абсолютно сухое вещество. Такие дрожжи хранятся 12 мес и более.
Определение трегалозы. Около 2 г сушеных дрожжей, взвешенных на аналитических весах, помещают в коническую колбу вместимостью 100 мл (одновременно с навеской для определения трегалозы берут навеску для определения влажности дрожжей), приливают 10,9 мл дистиллированной воды. После того как дрожжи набухнут, добавляют 39,1 мл 96%-ного этилового спирта (при этом концентрация спирта в колбе составит 75%). В случае, если исходный этиловый спирт имеет иную концентрацию (менее 96%), изменяют количество воды и спирта, чтобы раствор спирта при смешивании с дрожжами соответствовал 75%-ной концентрации.
К колбе присоединяют обратный холодильник, ставят ее в водяную баню с температурой 50 °С и выдерживают при этой температуре 30 мин. Затем фильтруют в другую чистую и сухую коническую колбу вместимостью 100 мл и промывают осадок 30-40 мл 75%-ного этилового спирта (несколько раз малыми количествами).
После этого колбу с фильтратом ставят в кипящую водяную баню до полного удаления растворителя. К полученной массе приливают 40 мл 1,3 н. раствора соляной кислоты и гидролизуют в кипящей бане в течение 4 ч с обратным водяным холодильником.
Содержимое колбы переносят в мерную колбу на 100 мл, гидролизат нейтрализуют 15%-ным раствором NaOH при индикаторе фенолфталеине, после чего добавляют для осветления 4 мл 20%-ного раствора сернокислого цинка и нейтрализуют насыщенным раствором гидрата окиси бария до темно-розового окрашивания. Раствор с образовавшимся в колбе осадком доводят до метки дистиллированной водой, хорошо перемешивают и фильтруют. В 20 мл фильтрата определяют сахар по Бертрану. Содержание трегалозы дается в пересчете на глюкозу. Расчет ведут на абсолютно сухое вещество дрожжей.
Пример. На определение взято 1,9269 г сушеных дрожжей влажностью 10% (с содержанием 90% СВ). На титрование 20 мл испытуемого раствора пошло 8,6 мл раствора KМnО (5 г KМnО в 1 л). 1 мл раствора KМnО соответствует 10,3 мг меди. Определяем количество меди в 20 мл испытуемого раствора
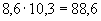 мг.
мг.
По табл.5 (см. приложение IV) находим соответствующее содержание сахара во взятой пробе (в мг):
88,2 мг меди - 46 мг сахара
88,6 мг меди -
=46,2 мг сахара в 20 мл испытуемого раствора.
46,2·5=231 мг сахара в 100 мл испытуемого раствора, т.е. в 1,9269 г дрожжей.
Определяем количество трегалозы в дрожжах в пересчете на воздушно-сухое вещество:
1,9269 г дрожжей - 0,231 мг сахара
100 г дрожжей -
=12,0% трегалозы.
Содержание трегалозы в пересчете на абсолютно сухое вещество:
90 г СВ - 12% трегалозы
100 г СВ -
=13,3% трегалозы на абсолютно сухое вещество.
Ниже приводится метод определения сахара по Бертрану.
Принцип метода. Сахар при кипячении со щелочным раствором окиси меди образует закись меди, которая восстанавливает окисное железо по уравнению
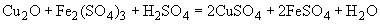 .
.
Количество восстановленного железа учитывается титрованием раствора марганцевокислого калия.
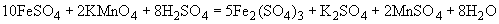 .
.
Титр раствора марганцевокислого калия устанавливают по щавелевой кислоте.
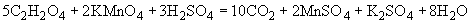 .
.
Ход определения. В коническую колбу вместимостью 100-150 мл пипеткой наливают 20 мл испытуемого нейтрального раствора сахара, полученного по методике, описанной выше. Содержание сахара в растворе не должно превышать 100 мг (оптимальное содержание сахара в растворе должно быть 50-80 мг). Затем в колбу приливают 40 мл свежеприготовленной смеси Фелинга, перемешивают и быстро нагревают до кипения. Кипятят 3 мин, избегая бурного кипения. Жидкость после кипения должна иметь синий цвет, который свидетельствует об избытке сернокислой меди. При отсутствии синей окраски (следовательно, было взято на определение больше 100 мг сахара) испытуемый нейтральный раствор сначала разбавляют, а затем определяют сахар и разбавление учитывают при расчете. Образовавшему красному осадку закиси меди дают отстояться, после чего синюю жидкость осторожно по палочке сливают на асбестовый фильтр трубки Аллина (рис.28), которую предварительно вставляют в колбу Бунзена, соединенную с водоструйным насосом. Осадок закиси меди стараются не переносить на фильтр, так как он на фильтре трудно растворяется. После того как синяя жидкость отфильтрована, оставшийся в колбе осадок закиси меди промывают горячей свежепрокипяченной водой, следя за тем, чтобы осадок находился все время под водой. Конец промывания устанавливают по исчезновению щелочной реакции на лакмус.
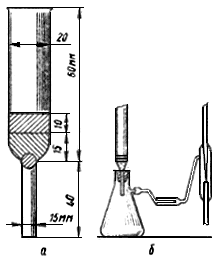
Рис.28. Прибор для определения сахара по Бертрану:
- трубка Аллина; - прибор для отсасывания фильтрата
Промытый осадок закиси меди растворяют в колбе раствором сернокислого окисного железа и серной кислоты, добавляя его небольшими порциями (например по 2-3 мл, затем, если не вся закись меди растворилась, добавляют еще 2-3 мл и т.д.).
После растворения закиси меди раствор сливают на асбестовый фильтр, на котором задержалось некоторое количество осадка, и разрыхляют верхний слой асбеста стеклянной палочкой, чтобы растворить всю закись меди и, присоединив водоструйный насос, фильтруют при небольшом разрежении. Колбу и фильтр промывают несколько раз небольшими порциями (по 2-3 мл) раствором сернокислого окисного железа и серной кислоты так, чтобы на фильтре совершенно не оставалось закиси меди. Затем во время фильтрования несколько раз промывают небольшими порциями холодной свежепрокипяченной воды, ополаскивая сначала колбу, а затем фильтр до исчезновения в промывных водах кислой реакции на лакмус.
Сине-зеленую жидкость, содержащую закисную соль железа, немедленно титруют из бюретки раствором KМnО до появления розового окрашивания.
На основании количества, пошедшего на титрование, KМnО (в мл) вычисляют отвечающее ему количество меди в миллиграммах и по табл.5 (см. приложение IV) находят соответствующее содержание сахара в миллиграммах.
Ускоренный метод ВНИИХП (по сумме кислот). Метод определения трегалозы в дрожжах является длительным и трудоемким, поэтому для оценки пригодности сушеных дрожжей к длительному хранению во ВНИИХПе разработан экспресс-метод, показывающий степень повреждения клеток высушенных дрожжей.
Метод заключается в следующем: из средней пробы отвешивают 0,5 г (на технических весах) высушенных дрожжей и помещают их в колбу Эрленмейера вместимостью 200 мл, туда же добавляют 100 мл дистиллированной воды комнатной температуры. Дрожжевая взвесь настаивается в течение 30 мин при комнатной температуре. Далее содержимое колбы титруют 0,1 н. раствором щелочи в присутствии индикатора фенолфталеина до слабо-розового окрашивания, затем туда же наливают 5 мл 40%-ного нейтрального формалина и снова титруют 0,1 н. раствором щелочи до слабо-розового окрашивания, появившегося после прибавления капли щелочи.
Общее количество 0,1 н. раствора щелочи, пошедшее на первое и второе титрование, является величиной суммы кислот.
Для экспресс-метода, определяемого по показателю "сумма кислот", разработаны абсолютные величины, позволяющие судить о пригодности сушеных дрожжей к хранению.
Ниже приводится зависимость величины суммы кислот от сроков хранения сушеных дрожжей.
|
Сумма кислот, мл 0,1 н. NaOH |
Срок хранения, мес |
|
До 1,1 |
Более 12 |
|
1,2-1,3 |
10-12 |
|
1,45-1,5 |
5 |
|
1,6-1,7 |
3 |
|
Более 1,7 |
Хранению не подлежат |
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЕ I
РАСТВОРЫ И РЕАКТИВЫ
Подготовка стеклянной посуды для проведения химических анализов
Стеклянную посуду, используемую для приготовления и отмеривания точных растворов, предварительно моют 1%-ным раствором двухромовокислого калия в концентрированной серной кислоте. Колбы и бюретки заполняют этим раствором и оставляют на сутки или несколько часов, пипетки на такое же время погружают в цилиндр, наполненный тем же раствором. После такой обработки посуду тщательно промывают водопроводной водой и ополаскивают дистиллированной водой.
Мерную стеклянную посуду (колбы, цилиндры, пипетки и бюретки) считают точной после проверки. Допустимые колебания вместимости не должны превышать 0,1%. Мерную посуду калибруют водой по массе при определенной температуре.
Масса разных объемов воды в граммах при температуре 16-24 °С приведена в табл.1.
Таблица 1
|
Температура воды, °С |
Масса разных объемов (в мм) воды, г |
||
|
1000 |
500 |
250 |
|
|
16 |
997,90 |
498,95 |
249,48 |
|
17 |
997,74 |
498,87 |
249,43 |
|
18 |
997,56 |
498,78 |
249,39 |
|
19 |
997,38 |
498,69 |
249,34 |
|
20 |
997,18 |
498,59 |
249,30 |
|
21 |
996,97 |
498,49 |
249,24 |
|
22 |
996,76 |
498,38 |
249,19 |
|
23 |
996,53 |
498,26 |
249,11 |
|
24 |
996,29 |
498,15 |
249,07 |
Если погрешность превышает допущенную при химических анализах, то производят калибровку мерной посуды.
Мерные колбы калибруют на вливание, а мерные цилиндры, бюретки и пипетки - на выливание; в первом случае посуда должна вмещать определенный объем, во втором случае этот объем должен вытекать из посуды.
При калибровке колб и мерных цилиндров воду взвешивают с точностью 0,1%, т.е. колбы вместимостью 1000 мл взвешивают с точностью до 1 г, а колбы вместимостью 100 мл - с точностью до 0,1 г.
Температура воды и колбы должна быть одинаковой, поэтому воду и колбы оставляют на 30 мин в одном и том же помещении.
Сухую чистую колбу или цилиндр взвешивают, затем определяют массу воды желаемого объекта при данной температуре и наливают в посуду до этой массы, метку ставят на горло колбы по нижнему краю мениска.
Мерные пипетки. При проверке пипетку заполняют до метки дистиллированной водой, затем выливают воду в заранее взвешенный сосуд и взвешивают с точностью в соответствии с вместимостью пипетки.
Так, при калибровке пипеток на 10 мл при температуре воды 18 °С сначала по табл.1 находят поправку. Масса 1000 мл при температуре 18 °С равна 997,56 г, следовательно, масса 10 мл воды при 18 °С составит 9,9756 г.
Погрузив нижний кончик пипетки в воду, всасывают ее несколько выше метки, затем, закрыв верхний конец пипетки указательным пальцем, вынимают пипетку, вытирают ее фильтровальной бумагой и дают жидкости стечь до метки, прикасаясь кончиком сосуда с водой. Затем содержимому пипетки дают стечь вдоль стенки тарированного стаканчика для взвешивания (бюксы) и, когда жидкость стечет, стаканчик закрывают и взвешивают. Если масса вытекшей воды больше или меньше 9,9756 г, то черту переносят соответственно выше или ниже и повторяют взвешивание до тех пор, пока разница не будет превышать 0±5 мг. После этого черту, нанесенную на пипетку, вытравливают плавиковой кислотой. Для этого на трубку пипетки предварительно наносят слой воска, а затем черту ножом. После этого снова проверяют пипетку путем взвешивания вылившейся жидкости (воды), операцию повторяют не менее 3 раз и среднюю величину считают правильной.
При калибровании бюреток и градуированных пипеток их устанавливают строго вертикально и наполняют водой на 5 мм выше нулевого уровня. Воду спускают точно до нулевого деления, удаляют каплю, висящую на кончике бюретки или пипетки, и при полностью открытом кране выливают непрерывной струей в тарированный стаканчик и затем взвешивают воду. Истечение жидкости при калибровании бюреток прерывают, когда уровень ее будет находиться приблизительно на 5 мм выше проверяемого деления. Через 30 с спускают воду точно до метки, удаляют каплю, свисающую с кончика бюретки, прикосновением его к стенке стакана и взвешивают вытекшую воду.
Например, чаще всего применяемую 50-миллиметровую бюретку проверяют в интервалах от 0 до 5 мл, от 0 до 10, от 0 до 15, от 0 до 20 и т.д. до 50 мл.
Получаемые при проверке бюретки отклонения от истинных объемов записывают и строят график следующим образом: по оси абсцисс откладывают объем бюретки в миллилитрах, а по оси ординат - отклонения от истинного объема. Если соединить полученные таким образом точки, то получают кривую, по которой легко определить соответствующую поправку для каждого отсчета бюретки.
Наполняют пипетку следующим образом: погрузив кончик пипетки в жидкость, засасывают ее ртом немного выше метки, быстро закрывают верхнее отверстие пипетки указательным пальцем и, осторожно приподнимая палец, устанавливают уровень жидкости по метке. Затем переносят пипетку в вертикальном положении к сосуду, в который должна быть налита вода, подняв палец, дают жидкости свободно стечь, после чего, выждав 15 с, касаются кончиком пипетки внутренней стенки сосуда. Не допускается выдувать раствор, оставшийся в оттянутом кончике пипетки.
Бюретки наполняют сверху через маленькую воронку с узким отверстием: открыв кран или зажим, заполняют раствором кончик бюретки, только после этого наполняют бюретку сначала до 1 см выше верхней метки, а затем устанавливают уровень жидкости точно по метке.
Растворы отмеривают из бюретки от нулевого деления, причем, установив уровень на 5 мм выше нужного деления, выжидают минутку и, приложив кончик бюретки к стенке сосуда, спускают жидкость точно до метки. При титровании отсчет производят через 1 мин после окончания титрования. Если титрование продолжается более 2 мин, выдержку можно не производить.
Стеклянные краны бюреток слегка смазывают вазелином. Для растворов едких щелочей применяют бюретки с зажимами, так как при хранении этих растворов в бюретках со стеклянными кранами происходит заедание кранов. Отсчет производят по нижнему краю мениска (при титровании темными растворами деления отсчитывают по верхнему краю мениска).
Для облегчения отсчетов пользуются кусочком белого картона, половина которого заклеена черной бумагой. Картон держат сзади бюретки так, чтобы граница между черным и белым участками находилась на 2 мм ниже уровня жидкости. В этом случае мениск кажется черным и резко выделяется на белом фоне.
Перекристаллизация веществ
В лаборатории довольно часто приходится очищать кристаллические вещества перекристаллизацией. Для этой цели растворитель нагревают до кипения, всыпают в него нужное количество веществ и полученный раствор отфильтровывают через воронку для горячего фильтрования, причем приемник (кристаллизатор, фарфоровую чашку, коническую колбу или стакан) помещают в охлаждающее вещество (холодная вода, снег или лед). Из ненасыщенного при высокой температуре раствора при охлаждении и постоянном помешивании стеклянной палочкой выделяется соль в виде мелких кристаллов. Их отделяют путем фильтрования при разрежении: маточный раствор упаривают до половины объема и снова выделяют новую порцию соли охлаждением, как это описано выше.
Величина отдельных кристаллов, выделившихся при перекристаллизации, зависит от скорости охлаждения раствора. Крупные кристаллы образуются при медленном охлаждении раствора, а мелкие - при быстром охлаждении его. Если при быстром охлаждении кристаллы не выделяются, то образование их можно вызвать, внося очень маленький кристаллик чистого вещества в раствор. При образовании мелких кристаллов вещество получается чище, так как в крупных кристаллах всегда содержится некоторое количество маточного раствора.
Для получения вещества в очень чистом виде перекристаллизацию производят несколько раз. Отфильтрованное вещество насыпают равномерным слоем на фильтровальную бумагу, сверху закрывают другим листом фильтровальной бумаги и сушат на воздухе. Вещества, не содержащие кристаллизационной воды, можно сушить в сушильном шкафу или эксикаторе.
Приготовление титрованных растворов
В 1 л нормального титрованного раствора содержится 1 г·экв данного вещества, т.е. количество граммов вещества, равное его молекулярной массе, деленной на число, соответствующее его основности. Например, для серной кислоты (HSО) 1 г·экв равен 98/2=49; 0,1 н. и 0,01 н. растворы содержат соответственно 0,1 или 0,01 г·экв вещества в 1 л.
Различные растворы неодинаково стойки: одни из них требуется проверять через каждые 5-10 дней, другие - через каждые 3 мес, третьи очень долго не изменяют своего титра. Причинами изменения титра служат поглощение углекислоты при соприкосновении растворов с кислородом воздуха, действие света, колебания температуры, влияние составных частей стекла. Точные растворы следует готовить из химически чистых реактивов хорошего качества, сохраняемых в хорошо закрытых банках. Бутыли с раствором щелочей и гипосульфита предохраняют от воздействия углекислоты воздуха при помощи хлоркальциевых трубок с натронной известью.
Титрованные растворы следует готовить путем точного отвешивания необходимого количества вещества и растворения его в точно определенном растворе воды. Обычно же растворы готовят с приблизительной точностью и вычисляют поправочный коэффициент, указывающий на отношение концентрации приготовленного раствора к теоретической. Например, если при нейтрализации 30 мл точно 0,1 н. раствора соляной кислоты приходится затратить не 50, а 49,5 мл раствора щелочи, то это значит, что приготовленная щелочь имеет несколько большую концентрацию, чем следует, и поправочный коэффициент будет равен 50/49,5=1,010. Затраченное количество данного раствора щелочи следует умножить на 1,010, чтобы получить количество точно 0,1 н. раствора.
Если при установлении поправки при трехкратном титровании расхождения равняются 0,05 мл, то производят еще одно титрование и вычисляют среднюю величину из всех титрований.
Свежепрокипяченную дистиллированную воду для приготовления титрованных растворов охлаждают и хранят в склянке, закрытой пробкой с вставленной в нее хлоркальциевой трубкой, содержащей натронную известь.
Титр 0,1 н. раствора щелочи устанавливают по 0,1 н. раствору серной кислоты, применяя при титровании фенолфталеин.
Растворы серной кислоты.
0,1 н. раствор. Грамм-эквивалент HSO равен 98/2=49, поэтому 0,1 н. раствор должен содержать в 1 л 4,9 г HSO.
Ареометром определяют плотность серной кислоты. Если она равна 1,84 (обычная продажная концентрированная кислота), то в 100 г кислоты содержится 95,6 г чистой серной кислоты (см. приложение IV, табл.5). Для приготовления 1 л раствора следует взять 4,90 г чистой серной кислоты, которые содержатся в 5,12 г данной кислоты. Вследствие неудобства взвешивания отмеривают пипеткой 2,78 мл кислоты (5,12/1,84), которые и соответствуют требуемому количеству (4,90 г) чистой кислоты. Это количество кислоты помещают в мерную литровую колбу с дистиллированной водой, раствор перемешивают и доливают колбу водой до метки. Раствор, полученный таким образом, не является точно децинормальным; титр его может быть установлен различными способами, например при помощи NaСО. Несколько граммов химически чистого углекислого натрия нагревают на песчаной бане в течение 3 мин при 270-300 °С, после охлаждения в эксикаторе отвешивают 1,325 г и растворяют их в 250 мл дистиллированной воды, получив таким образом точно 0,1 н. раствор соды.
Для установления титра раствора серной кислоты наливают его в бюретку; в колбу вместимостью около 100 мл пипеткой отмеривают 25 мл раствора соды, прибавляют 1-2 капли метилового оранжевого (1%-ный водный раствор) и титруют раствор соды раствором серной кислоты до перехода желтой окраски в оранжево-розовую. Если на титрование израсходовано 24 мл раствора серной кислоты, то поправочный коэффициент равняется 25/24=1,0416; это указывает, что раствор серной кислоты крепче 0,1 н. в 1,0416 раза.
Титр раствора серной кислоты можно также установить по буре (борнокислый натрий NaBO·10НО). Грамм-эквивалент буры равен половине ее молекулярной массы.
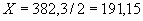 г.
г.
Буру надо предварительно перекристаллизовать, причем она кристаллизуется с десятью молекулами воды только при температуре ниже 60 °С, выше этой температуры образуется октаэдрическая бура с пятью молекулами воды (NaBO·5НО). Поэтому если при фильтровании горячего раствора буры выпадают кристаллы, то их надо снова растворить при нагревании, затем осторожно охладить раствор, не давая кристаллизоваться, и только тогда при быстром помешивании палочкой вызвать образование кристаллов NaBО·10НО. Буру сушат между листами фильтровальной бумаги на воздухе. Титр раствора серной кислоты устанавливают при помощи отдельных навесок буры, растворяя их в дистиллированной воде и целиком титруя раствором кислоты с метиловым оранжевым. Следует произвести три определения и взять среднее значение.
Пример. Взято 0,4020 г буры. Для нейтрализации потребовалось 20,3 мл раствора кислоты, 20,3 мл точно 0,1 н. раствора HSO должны соответствовать 0,019115·20,3=0,3880 г буры. В 20,3 мл взятого раствора HSО содержится буры 0,4020 г. Следовательно, отношение нашего раствора к точно 0,1 н. равно 0,4020/0,3880=1,0361; эта величина представляет собой поправку для данного раствора серной кислоты.
Нормальный раствор. Этот раствор готовят так же, как 0,1 н., но количество серной кислоты берут в 10 раз больше.
Навеску соды для установки титра берут тоже в 10 раз больше, т.е. 13,25 г. Можно установить титр кислоты и по раствору щелочи, соответствующей нормальности с известным титром.
0,5 н. раствор. Его готовят путем разбавления водой 1 н. раствора в 2 раза.
Растворы соляной кислоты. Нормальный раствор. Грамм-эквивалент НCl равен 36,46; в 1 мл раствора должно содержаться 0,03646 г НCl.
Концентрированная кислота относительной плотностью 1,19 (см. приложение III, табл.5) содержит 37,23% НCl, поэтому на 1 л нормального раствора ее нужно взять 97,93 г или 82,3 мл.
83 мл концентрированной соляной кислоты плотностью 1,19 разбавляют водой до объема 1 л.
Титр раствора устанавливают по NaCO. Для этого 5 г карбоната натрия взвешивают в платиновой чашке, нагревают до постоянной массы при 280-300 °С на песчаной бане, помешивая платиновой проволочкой, и охлаждают. Точную навеску около 1 г NaCO, полученную описанным способом, растворяют в 100 мл воды, приливают 2 капли метилового оранжевого и титруют раствором соляной кислоты до появления розовато-оранжевого окрашивания. Поправочный коэффициент вычисляют по формуле
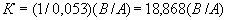 ,
,
где - количество раствора соляной кислоты, пошедшее на титрование, мл; - навеска карбоната натрия, г.
0,1 н. раствор. 100 мл нормального раствора соляной кислоты разбавляют водой до 1 л.
Растворы гидроксида натрия. 0,1 н. раствор. Отвешивают около 5 г продажного препарата гидроксида натрия, быстро ополаскивают его дистиллированной водой, переносят в мерную колбу вместимостью 1 л, растворяют в небольшом количестве воды и доливают раствор до метки водой. Титр полученного раствора устанавливают по 0,1 н. раствору серной кислоты определенного титра или по янтарной кислоте. Для этого взвешивают 1,4755 г перекристаллизованной янтарной кислоты, которую растворяют водой в мерной колбе вместимостью 250 мл, получая таким образом 0,1 н. раствор (грамм-эквивалент янтарной кислоты равен 118,05/2). К 25 мл этого раствора добавляют 1-2 капли фенолфталеина и титруют раствором гидроксида натрия, приливая его из бюретки до появления остающегося слабо-розового окрашивания.
Поправку вычисляют так же, как указано выше, при приготовлении 0,1 н. раствора серной кислоты.
При приготовлении раствора щелочи следует применять дистиллированную воду, предварительно освобожденную от углекислоты кипячением в течение 3 мин. Вместо этого раствор приготовленной щелочи можно обработать небольшим количеством хлористого бария, который с имеющейся в растворе углекислотой образует нерастворимую соль, выпадающую в осадок.
Раствор гипосульфита.
Грамм-эквивалент NaSО·5НО равен 248. Для приготовления 0,1 н. раствора гипосульфита 24,8 г кристаллической соли NaSO·5НО растворяют в 1 л дистиллированной воды, прокипяченной для удаления углекислоты и затем охлажденной. Бутыль с раствором гипосульфита рекомендуется покрасить темным лаком или обернуть темной бумагой для защиты раствора от действия света.
Титр приготовленного раствора гипосульфита устанавливают по двухромовокислому калию, предварительно 2-3 раза перекристаллизованному и высушенному до постоянной массы при 130 °С.
Для получения точно 0,1 н. раствора KСrО отвешивают на аналитических весах 4,9037 г двухромовокислого калия и растворяют их в 1 л воды. В коническую колбу вносят около 20-40 мл 2 н. раствора серной кислоты, 20-40 мл 0,2 н. раствора йодистого калия и точно отмеренное количество 0,1 н. раствора KСrО. Колбу закрывают часовым стеклом, оставляют стоять 3 мин, добавляют 200-300 мл дистиллированной воды и титруют раствором гипосульфита до светло-желтой окраски, затем добавляют несколько капель 1%-ного раствора крахмала (раствор окрашивается в синий цвет) и дотитровывают до полного исчезновения синей окраски.
Пример. 25 мл 0,1 н. раствора KСrО соответствуют 23,8 мл гипосульфита. Поправка 0,1 н. раствора гипосульфита составит 25,0/23,80=1,0504.
Титр раствора гипосульфита устанавливают обычно через две недели после приготовления, так как титр его меняется вследствие разлагающего действия углекислоты, находящейся в дистиллированной воде.
0,01 н. раствор гипосульфита готовят путем разбавления 100 мл 0,1 н. раствора NaSO водой в мерной колбе вместимостью 1 л. Раствор доводят до метки дистиллированной водой, не содержащей углекислоты.
0,2 н. раствор йодистого калия. 16,6 г KI помещают в мерную колбу вместимостью 500 мл, растворяют в небольшом количестве дистиллированной воды и доводят раствор водой до метки.
0,1 н. раствор йода. Грамм-эквивалент йода равен 126,93; в 0,1 н. растворе должно содержаться 12,693 г йода в 1 л раствора.
Продажный йод очищают возгонкой. Так как йод часто загрязнен Cl и Вr, то перед возгонкой его растирают в агатовой ступке с несколькими кристалликами йодистого калия. При возгонке Сl и Вr переходят в соединение с калием. На дно стаканчика вместимостью 50-100 мл без носика насыпают кристаллы йода. Сверху стаканчика помещают небольшую круглодонную колбу, в которую наливают воду для охлаждения.
Стаканчик с йодом помещают в асбестовую сетку или на песчаную баню и осторожно нагревают. Если йод не успевает охладиться на дне колбы, то пары его улетучиваются в воздух. В этом случае нужно ослабить нагревание. При соблюдении указанных условий дно колбы покрывается красивыми кристаллами йода, которые переносят в стаканчик со стеклянной крышкой.
Рекомендуется растворять йод в небольшом объеме очень концентрированного раствора йодистого калия и только после этого разбавлять водой до нужного объема. Быстрое растворение йода основано на образовании комплексного, легко растворимого в воде соединения K+I=KI, образование которого не мешает титрованию.
25 г химически чистого йодистого калия растворяют в возможно малом количестве воды в мерной колбе вместимостью 1 л, прибавляют 12,7 г йода, очищенного возгонкой, и взбалтывают до полного растворения его, после чего доводят раствор до метки. Раствор сохраняют в склянке оранжевого стекла с притертой пробкой. Титр устанавливают по гипосульфиту, для чего 20 мл раствора йода титруют 0,1 н. раствором гипосульфита до тех пор, пока цвет раствора не станет светло-желтым, затем прибавляют 2-3 капли раствора крахмала и дотитровывают до полного обесцвечивания раствора.
Пример. 20 мл йодного раствора эквивалентны 21,3 мл 0,1 н. раствора NaSO с поправкой гипосульфита 0,98, что соответствует 21,3·0,98=20,87 мл точно 0,1 н. раствора. 20 мл раствора йода соответствуют 20,87 мл 0,1 н. раствора гипосульфита. Поправка раствора йода составит 20,87/20=1,0435.
0,1 н. раствор уксусной кислоты.
Грамм-эквивалент СНСООН равен 60,03; для приготовления 0,1 н. раствора следует взять 60,03/10=6,003 г уксусной кислоты.
Из концентрированной кислоты в зависимости от процентного содержания в ней СНСООН готовят 0,1 н. раствор.
Пример. Имеется 80%-ный раствор уксусной кислоты; для приготовления 0,1 н. раствора нужно взять 6·100/80=7,5 г кислоты
Принимая плотность уксусной кислоты за 1, отмериваем 7,5 мл и доводим их дистиллированной водой до объема 1 л.
0,1 н. раствор двухромовокислого калия. Для приготовления 1 н. раствора двухромовокислого калия величину навески находят из уравнения
 .
.
Одна молекула KСrО соответствует 6 атомам йода, поэтому нормальный раствор должен содержать в 1 л 294,21/6=49,035 г KСrО.
Для приготовления 0,1 н. раствора KСrО берут навеску 4,9035 г KСrО, перекристаллизованного и высушенного до постоянной массы при 120-130 °С, растворяют в дистиллированной воде и доводят объем до 1 л. В 1 мл раствора содержится 4,9035 г KСrО, что соответствует 12,69 мг йода или 0,8 мл кислорода.
Приготовление буферных растворов
Тетраборнокислый натрий
NaBO·10НО (бура). Подготовка вещества. 100 г препарата растворяют в 550 мл воды при температуре 50-60 °С (при более высокой температуре кристаллизуется NaBO·5НО). Растворы фильтруют и охлаждают до 25-30 °С. При последующем энергичном помешивании раствора происходит кристаллизация буры. Образующиеся кристаллы отфильтровывают через отсасывающую воронку, снова подвергают растворению и кристаллизации. Затем кристаллы сушат между листами фильтровальной бумаги до тех пор, пока они не перестанут прилипать к стеклянной палочке.
Приготовление раствора. Для приготовления 0,05 М раствора тетра-борнокислого натрия 19,10 г полученного препарата растворяют в воде и доводят объем раствора водой до 1 л.
Янтарная кислота
СНО. Подготовка вещества. 100 г препарата растворяют при кипячении в 165 мл воды, раствор фильтруют через воронку с обогревом, фильтрат постоянно помешивают, после охлаждения раствора кристаллы отфильтровывают на отсасывающей воронке и снова подвергают растворению и кристаллизации. Затем кристаллы сушат между листами фильтровальной бумаги до тех пор, пока они не перестанут прилипать к стеклянной палочке.
Приготовление раствора. Для приготовления 0,05 М раствора янтарной кислоты 5,90 г полученного препарата растворяют в воде и доводят объем раствора водой до 1 л.
Калийбифталат
С НОK. Подготовка вещества. 50 г препарата растворяют в 100 мл воды, кристаллизацию ведут при температуре не ниже 25 °С, так как при более низкой температуре образуются кристаллы трифталата калия - более кислой соли. Полученные кристаллы сушат до постоянной массы при температуре 110-115 °С.
НОK. Подготовка вещества. 50 г препарата растворяют в 100 мл воды, кристаллизацию ведут при температуре не ниже 25 °С, так как при более низкой температуре образуются кристаллы трифталата калия - более кислой соли. Полученные кристаллы сушат до постоянной массы при температуре 110-115 °С.
Приготовление раствора. Для приготовления 0,2 М раствора бифталата калия 40,84 г полученного препарата растворяют в воде и объем раствора доводят водой до 1 л.
Калий хлористый KCl. Подготовка вещества. Препарат ч.д.а. 3-4 раза перекристаллизовывают из воды. 350 г препарата растворяют в 650 мл воды, раствор нагревают до кипения, фильтруют и охлаждают до 25 °С, при охлаждении из раствора выпадает около 100 г хлористого калия. Кристаллы сушат в течение 48 ч при температуре 115-120 °С.
Приготовление раствора. Для приготовления 0,2 М раствора хлористого калия 14,91 г препарата растворяют в воде и объем препарата доводят водой до 1 л.
Калий фосфорнокислый однозамещенный
KНРО. Подготовка вещества. Калий фосфорнокислый однозамещенный 2 раза перекристаллизовывают из воды. 100 г препарата растворяют при кипячении в 150 мл воды. Раствор отфильтровывают на отсасывающей воронке и снова подвергают растворению и кристаллизации. Кристаллы сушат до постоянной массы при температуре 110 °С. Потери полученного препарата при прокаливании составляют 13,23±0,01%.
Приготовление раствора. Для приготовления 0,1 н. раствора фосфорнокислого калия 13,62 г полученного препарата растворяют в воде и доводят объем раствора водой до 1 л.
Натрий углекислый безводный
NaCO. Подготовка вещества. 40 г препарата нагревают в платиновом тигле на песочной бане при температуре 270-300 °С до постоянной массы Во время нагревания препарат часто перемешивают платиновым шпателем или проволокой (которые оставляют в тигле во время нагревания) и охлаждают в эксикаторе. Полученный безводный углекислый натрий ввиду его гигроскопичности сохраняют в банках с притертыми пробками.
Приготовление раствора. Для приготовления 0,1 н. раствора углекислого натрия 5,30 г полученного препарата растворяют в воде и объем раствора доводят водой до 1 л.
Двузамещенный фосфат натрия Na
НРО
·12Н
О.
Подготовка вещества. 150 г двузамещенного двенадцативодного фосфата натрия (х.ч.) NaHPO·12НО дважды перекристаллизовывают из воды. Перекристаллизованный препарат нагревают в фарфоровой чашке на водяной бане при непрерывном перемешивании до полного высыхания препарата. Полученную соль (NaHPO·12НО) высушивают над расплавленным хлористым кальцием до постоянной массы. Потери препарата при прокаливании составляют 25,28±0,01%.
Приготовление раствора. Для приготовления 0,1 н. раствора двузамещенного фосфата натрия 17,81 г полученного препарата растворяют в воде и объем раствора доводят водой до 1 л.
Гидроксид натрия. Подготовка вещества и приготовление раствора. 250 г препарата растворяют в 300 мл воды в стеклянном цилиндре с пробкой. Раствор хорошо перемешивают, закрывают цилиндр пробкой и оставляют в покое на несколько дней, в течение которых углекислый натрий, нерастворимый в щелочи указанной концентрации, оседает на дно цилиндра. После этого главную массу прозрачного раствора сливают и соответствующим разбавлением полученного раствора водой, не содержащей углекислоты, приготовляют растворы гидроксида натрия необходимой концентрации (0,1 н. и 1 н.).
Примечание. Навески препаратов берут с точностью до 0,01 г.
Буферные смеси, применяемые для определения рН. Буферные смеси, применяемые для определения интервала перехода рН от 0,13 до 5,8, приведены в табл.2; для определения интервала перехода рН от 6 до 11 - в табл.3 и для определения интервала перехода рН от 11,2 до 14 - в табл.4.
Таблица 2
|
рН |
Коли- |
Соляная кислота |
0,2 М раствор хлористого калия, мл |
0,2 М раствор бифталата калия, мл |
0,05 М раствор янтарной кислоты, мл |
0,05 М |
|
|
1 н. раствора, мл |
0,2 н. раствора, мл |
||||||
|
0,13 |
- |
100 |
- |
- |
- |
||
|
0,28 |
40 |
60 |
- |
- |
- |
||
|
0,74 |
80 |
20 |
- |
- |
- |
||
|
1,0 |
26,5 |
- |
48,5 |
25 |
- |
||
|
1,2 |
42,75 |
- |
32,25 |
25 |
- |
||
|
1,4 |
54,25 |
- |
20,75 |
25 |
- |
||
|
1,6 |
61,85 |
- |
13,15 |
25 |
- |
||
|
1,8 |
66,7 |
- |
8,3 |
25 |
- |
||
|
2,0 |
69,7 |
- |
5,3 |
25 |
- |
||
|
2,2 |
71,65 |
- |
3,35 |
25 |
- |
||
|
2,2 |
51,65 |
- |
23,35 |
- |
25 |
||
|
2,4 |
55,2 |
- |
19,8 |
- |
25 |
||
|
2,6 |
58,52 |
- |
16,48 |
- |
25 |
||
|
2,8 |
61,79 |
- |
13,21 |
- |
25 |
- |
- |
|
3,0 |
64,84 |
- |
10,16 |
- |
25 |
- |
- |
|
3,2 |
67,65 |
- |
7,35 |
- |
25 |
- |
- |
|
3,4 |
70,05 |
- |
4,95 |
- |
25 |
- |
- |
|
3,0 |
- |
- |
- |
- |
- |
98,6 |
1,4 |
|
3,2 |
- |
- |
- |
- |
- |
96,5 |
3,5 |
|
3,4 |
- |
- |
- |
- |
- |
94,0 |
6,0 |
|
3,6 |
- |
- |
- |
- |
- |
90,5 |
9,5 |
|
3,8 |
- |
- |
- |
- |
- |
86,3 |
13,7 |
|
4,0 |
- |
- |
- |
- |
- |
82,2 |
17,8 |
|
4,2 |
- |
- |
- |
- |
- |
77,8 |
22,2 |
|
4,4 |
- |
- |
- |
- |
- |
73,8 |
26,2 |
|
4,6 |
- |
- |
- |
- |
- |
70,0 |
30,0 |
|
4,8 |
- |
- |
- |
- |
- |
66,5 |
33,5 |
|
5,0 |
- |
- |
- |
- |
- |
63,2 |
36,8 |
|
5,2 |
- |
- |
- |
- |
- |
60,5 |
39,5 |
|
5,4 |
- |
- |
- |
- |
- |
57,9 |
42,1 |
|
5,6 |
- |
- |
- |
- |
- |
55,7 |
44,3 |
|
5,8 |
- |
- |
- |
- |
- |
54,0 |
46,0 |
Таблица 3
|
рН |
0,05 M раствор буры, мл |
0,1 М раствор однозамещенного фосфата калия, мл |
0,1 М раствор углекислого натрия, мл |
|
6,0 |
12,3 |
87,7 |
- |
|
6,2 |
17,0 |
83,0 |
- |
|
6,4 |
23,0 |
77,0 |
- |
|
6,6 |
28,8 |
71,2 |
- |
|
6,8 |
34,2 |
65,8 |
- |
|
7,0 |
39,0 |
61,0 |
- |
|
7,2 |
43,4 |
56,6 |
- |
|
7,4 |
46,4 |
53,6 |
- |
|
7,6 |
49,2 |
50,8 |
- |
|
7,8 |
52,0 |
48,0 |
- |
|
8,0 |
55,0 |
45,0 |
- |
|
8,2 |
57,6 |
42,4 |
- |
|
8,4 |
62,0 |
38,0 |
- |
|
8,6 |
63,0 |
32,0 |
- |
|
8,8 |
75,2 |
24,8 |
- |
|
9,0 |
86,8 |
13,2 |
- |
|
9,2 |
100,0 |
- |
- |
|
9,4 |
64,3 |
- |
35,7 |
|
9,6 |
44,5 |
- |
55,5 |
|
9,8 |
33,3 |
- |
66,7 |
|
10,0 |
24,6 |
- |
75,4 |
|
10,2 |
17,85 |
- |
82,15 |
|
10,4 |
13,1 |
- |
86,9 |
|
10,6 |
8,5 |
- |
91,5 |
|
10,8 |
5,25 |
- |
94,75 |
|
11,0 |
2,7 |
- |
97,3 |
Таблица 4
|
Количество раствора, мл |
рН смеси |
|
|
I |
II |
|
|
10,0 |
0 |
8,30 |
|
9,75 |
0,25 |
8,03 |
|
9,50 |
0,5 |
7,86 |
|
9,0 |
1,0 |
7,65 |
|
8,0 |
2,0 |
7,35 |
|
7,0 |
3,0 |
7,35 |
|
6,0 |
4,0 |
6,98 |
|
5,0 |
5,0 |
6,81 |
|
4,0 |
6,0 |
6,64 |
|
3,0 |
7,0 |
6,45 |
|
2,0 |
8,0 |
6,24 |
|
1,0 |
9,0 |
5,91 |
|
0,5 |
9,5 |
5,60 |
|
0,25 |
9,75 |
4,98 |
|
0 |
10,0 |
4,53 |
Общий объем смеси равен 100 мл. Растворы отмеривают бюреткой.
Индикаторы для определения кислотности и щелочности. Интервал рН перехода окраски индикатора определяют буферными смесями.
Шкала буферных смесей должна быть составлена так, чтобы для индикатора с двумя приведенными в табл.2 значениями перехода можно было отчетливо обнаружить оба перехода, для чего каждому из двух приведенных в этой таблице значений рН предшествуют две буферные смеси, отличающиеся одна от другой на 0,2 рН.
Испытание фенолфталеина проводят в интервале перехода рН от 8,2 до 10, что соответствует изменению окраски от бесцветной к красной.
Первое изменение окраски от бесцветной к красной должно наблюдаться в смеси со значением рН 8,2 (по сравнению со смесями рН 7,8-8) и должно нарастать в смесях с рН 8,4-8,6. Смесь со значением рН 10 должна быть моментом прекращения нарастания интенсивности окраски. Две предшествующие смеси с рН 9,6-9,8 обнаруживают еще усиление окраски; сравнение же с двумя последующими смесями с рН 10,2 и 10,4 обнаруживает прекращение нарастания интенсивности окраски.
Таким образом, для испытаний одноцветных индикаторов шкалы буферных смесей построены то тому же принципу.
Шкала для испытаний фенолфталеина состоит из буферных смесей со следующими значениями рН: 7,8; 8; 8,2; 8,4; 8,6; 9,6; 9,8; 10; 10,2 и 10,4.
Для испытания индикатора метилового фиолетового составляют шкалу буферных смесей, соответствующую значениям рН: 0,13; 0,28; 0,74; 1; 1,4; 1,8; 2,2; 2,6; 3; 3,4.
За изменением окраски ведут наблюдение тотчас же по прибавлении во все пробирки раствора индикатора. Для испытания в интервале рН 3-3,4 необходимо применять бифталатные буферные смеси.
Для приготовления шкалы наливают по 5 мл буферных смесей (соответствующих значениям рН, приведенным в табл.2 с двумя предшествующими и двумя последующими смесями, отличающимися одна от другой на 0,2 рН) в 10 хорошо вымытых, пропаренных, сухих пробирок бесцветного стекла и одинакового диаметра.
Фосфатные буферные смеси. Буферные смеси с концентрацией водородных ионов от 8,3 до 4,5. Приготавливают растворы 1/15 молярных фосфатов.
I. Двузамещенный фосфорнокислый натрий (кристаллический NaHPO·12НО) - 23,881 г в 1 л.
II. Однозамещенный фосфорнокислый калий (KНРО) - 9,468 г в 1 л.
Растворы фосфатов I и II смешивают в разных соотношениях (табл.4).
Буферные смеси с концентрацией водородных ионов от 4,96 до 1,04. Приготовляют раствор цитрата.
I. Кристаллическая лимонная кислота 21,008 г +400 мл 0,05 н. гидроксида натрия в 1 л.
II. 0,1 н. раствор соляной кислоты.
При смешивании растворов в разных соотношениях получают следующие рН смесей (табл.5).
Таблица 5
|
Количество растворов, мл |
рН смеси |
|
|
Цитрат (I) |
0,1 н. НСl (II) |
|
|
10,0 |
0 |
4,96 |
|
9,0 |
1,0 |
4,83 |
|
8,0 |
2,0 |
4,65 |
|
7,0 |
3,0 |
4,45 |
|
6,0 |
4,0 |
4,19 |
|
5,0 |
5,0 |
3,69 |
|
4,5 |
5,5 |
3,37 |
|
4,0 |
6,0 |
2,98 |
|
3,0 |
7,0 |
1,92 |
|
2,0 |
8,0 |
1,42 |
|
1,0 |
9,0 |
1,17 |
|
0 |
10,0 |
1,04 |
Индикаторы, наиболее применяемые при контроле дрожжевого производства, и методы их приготовления
Бромтимол синий. 0,2 г индикатора растворяют в 50 мл 70%-ного этилового спирта; 10 мл спиртового раствора разбавляют дистиллированной водой до 100 мл.
1%-ный раствор фенолфталеина. 1 г фенолфталеина растворяют в 60 мл 96%-ного этилового спирта. Раствор доводят до объема 100 мл дистиллированной водой; на 100 мл титруемого раствора берут не более 2 капель индикатора.
0,1%-ный раствор метилового оранжевого. 0,1 г метилового оранжевого растворяют в 100 мл воды. При титровании на 25 мл раствора берут не более 3 капель индикатора.
0,02%-ный раствор метилового красного. 0,2 г индикатора растворяют в 100 мл горячей воды, охлаждают и фильтруют. На каждые 100 мл титруемого раствора берут 2-3 капли раствора индикатора.
Смешанный индикатор. Готовят спиртовые растворы следующих индикаторов: метиловый красный - 0,2 г на 100 мл 96%-ного спирта, метиловый синий - 0,1 г на 100 мл спирта; оба раствора смешивают в соотношении 1:1.
Универсальный индикатор. Готовят спиртовые растворы следующих индикаторов (0,1%-ные растворы в 96%-ном этиловом спирте) и смешивают их в определенных количествах: метиловый оранжевый - 15 мл, метиловый красный - 5 мл, бромтимоловый синий - 20 мл, фенолфталеин - 20 мл, тимолфталеин - 20 мл.
Раствор лакмуса и лакмусовая бумага. Лакмус растирают в фарфоровой ступке в порошок, помещают в фарфоровую чашку, заливают 90%-ным этиловым спиртом и нагревают на кипящей водяной бане 15 мин. Раствор сливают, операцию повторяют еще раз с новой порцией спирта и вновь сливают раствор. Затем в чашку с лакмусом наливают горячую дистиллированную воду, переводят содержимое чашки в высокий цилиндр и в течение нескольких часов периодически взбалтывают. Раствор оставляют для отстаивания в течение 10-12 ч, после чего жидкость сливают с осадка, сгущают ее на водяной бане до 1/3 объема, пересыщают уксусной кислотой и вновь сгущают до консистенции сиропа. Затем еще раз обрабатывают 96%-ным спиртом для осаждения синего красящего вещества, являющегося индикатором.
Осадок отфильтровывают и растворяют в таком количестве теплой дистиллированной воды, чтобы 3 капли полученного раствора давали заметное окрашивание в 50 мл дистиллированной воды.
В чашку с раствором погружают полоски бумаги размером 10х25 см и, вынув их из раствора, при помощи толстой стеклянной палочки выравнивают слой краски на бумаге. Глянцевую бумагу применять не рекомендуется из-за неравномерности получаемой окраски. Бумагу развешивают, сушат в комнате, где нет кислотных или аммиачных паров, а затем режут на мелкие полоски.
1%-ный раствор крахмала. 1 г растворимого крахмала размешивают в 3-5 мл дистиллированной воды; полученную смесь вливают в кипящую воду (95-97 мл), нагревают до просветления раствора, охлаждают и фильтруют.
Дифениламин. 1 г кристаллического препарата растворяют в 100 мл химически чистой серной кислоты относительной плотностью 1,84.
Физические и химические показатели описанных выше индикаторов, применяемых для определения кислотности и щелочности, приведены в табл.6 (справочная).
Таблица 6
|
Индикатор |
Свойства |
Молеку- |
Интервал перехода окраски |
Способ приготовления растворов индикаторов |
Количество |
|
|
рН |
цвет |
|||||
|
Бромкрезол пурпуровый (бромкрезолпурпур). Диброморто- |
Бесцветный мелко- |
540,23 |
5,2-6,8 |
От желтой к пурпурной |
0,1 г препарата растирают в фарфоровой ступке с 9,25 мл 0,02 н. раствора гидроксида натрия и по растворении препарата доводят объем раствора водой до 250 мл |
0,05 |
|
Бромтимоловый синий (бромтимолблау) 3,3- дибромтимолсульфон- |
Бесцветный мелко- |
624,39 |
6-7,6 |
От желтой к синей |
0,1 г препарата растирают в фарфоровой ступке с 8 мл 0,02 н. раствора гидроксида натрия и по растворении препарата доводят объем раствора водой до 250 мл |
0,1 |
|
Бромфеноловый синий (бромфенолблау) 3,3-5,5-тетрабром- |
Мелкие бесцветные кристаллы, допустима серовато-розовая или желтоватая окраска |
669,99 |
3,0-4,6 |
То же |
0,1 г препарата растирают в фарфоровой ступке с 7,5 мл 0,02 н. раствора гидроксида натрия и по растворении препарата доводят объем водой до 250 мл |
0,5 |
|
Крезоловый красный (крезолрот). Орто- |
Мелкокристал- |
382,41 |
7,2-8,8 |
От желтой к малиново- |
0,1 г препарата растирают в фарфоровой ступке с 13,1 мл 0,02 н. раствора гидроксида натрия и по растворении препарата доводят объем раствора водой до 250 мл |
0,05 |
|
Метиловый красный (метилрот). 4-диметиламиноазо- |
Блестящие фиолетовые кристаллы или красно-бурый порошок |
269,29 |
4,2-6,2 |
От красной к желтой |
0,1 г препарата растирают в фарфоровой ступке с 18,6 мл 0,02 н. раствора гидроксида натрия и по растворении препарата доводят объем раствора водой до 250 мл |
0,05 |
|
Метиловый оранжевый. Метилоранж 4-ди- |
Оранжевый кристаллический порошок, допустим коричневый оттенок |
327,33 |
3-4,4 |
От красной к желтой |
0,1 г препарата растворяют в 80 мл горячей воды и по охлаждении доводят водой до 100 мл |
0,05 |
|
Метиловый фиолетовый (метилвиолет). Пентаметилпараро- |
Зелень с металлическим блеском кристаллический порошок |
393,95 |
0,13-3,2 |
От желтой (рН 0,13) через зеленую и голубую к синей (около рН 1,5) и далее до фиолетовой (рН 2,6-3,2) |
0,5 г препарата растворяют в 100 мл воды |
0,1 |
|
Нейтральный красный (нейтральрот). 3- |
Черный или черно- |
288,77 |
6,8-8 |
От красной к желтой |
0,1 г препарата растворяют в 70 мл 95%-ного спирта и доводят объем раствора водой до 100 мл |
0,05 |
|
Тимоловый синий (тимолблау). Тимолсульфон- |
Кристаллический порошок от темно- |
466,57 |
1,2-2,8 |
То же |
0,1 г препарата растирают в фарфоровой ступке с 10,75 мл 0,02 н. раствора гидроксида натрия; после растворения препарата доводят объем раствора водой до 100 мл (раствор А); 10 мл раствора А разбавляют водой до 100 мл (раствор Б) |
0,15 |
|
Тимолфталеин. 2,2-диметил- |
Белый кристаллический порошок |
430,52 |
9,4-10,6 |
От бесцветной к синей |
0,1 г препарата растворяют в 100 мл 95%-ного спирта |
0,05 |
|
Феноловый красный (фенолрот). Фенолсуль- |
Темно-красный мелкий кристаллический порошок |
354,36 |
6,8-8,4 |
От желтой к красной |
0,1 г препарата растирают в фарфоровой ступке с 14,2 мл 0,02 н. раствора гидроксида натрия и по растворении препарата доводят объем раствора водой до 250 мл |
0,05 |
|
Фенолфталеин |
Белый или бледно-желтоватый кристаллический порошок |
318,31 |
8,2-10 |
От бесцветной к красной |
0,1 г препарата растворяют в 100 мл 95%-ного спирта |
0,05 |
ПРИЛОЖЕНИЕ II
ПРИГОТОВЛЕНИЕ РАСТВОРОВ ДЛЯ КОНТРОЛЯ ПРОИЗВОДСТВА
Растворы для определения инвертного сахара по методу Офнера
Навеску 5 г перекристаллизованной сернокислой меди, взвешенной в стакане вместимостью 100 мл, растворяют в 50-60 мл дистиллированной воды, затем переводят в мерную колбу вместимостью 1000 мл.
В отдельном стакане смешивают навески: 300 г мелкокристаллического виннокислого калия-натрия, 10 г безводного углекислого натрия и 50 г двузамещенного фосфорнокислого натрия, приливают 500 мл дистиллированной воды, подогретой до 50 °С. Для ускорения растворения смеси солей перемешивают содержимое стакана стеклянной палочкой. Полученный раствор переводят в мерную колбу с раствором сернокислой меди, перемешивают и фильтруют через двойной фильтр. Раствор должен быть совершенно прозрачным. Раствор хранят в стеклянной посуде из темного стекла с притертой пробкой.
Примечание. Сернокислая медь (CuSО), применяемая для приготовления раствора Офнера, не должна быть загрязнена соединениями железа. В случае загрязнения этими соединениями реактив следует перекристаллизовать с добавлением соляной кислоты. Наличие железа в CuSO устанавливают следующим образом: пять частей CuSO растворяют в воде, добавляют несколько капель концентрированной азотной кислоты и кипятят. По охлаждении насыщают раствор аммиаком до тех пор, пока вся медь не перейдет в синий раствор, затем фильтруют и промывают разведенным аммиаком. Железо остается на фильтре в виде Fе(ОН).
0,0323 н. раствор двуххромовокислого калия. Навеску 1,5839 г перекристаллизованного и высушенного при 130-150 °С двухромовокислого калия, взвешенного в стеклянном стакане, переводят дистиллированной водой в мерную колбу вместимостью 1 л, доводят содержимое колбы до метки и перемешивают.
0,5%-ный раствор крахмала. 0,5 г взвешенного в стакане вместимостью 50 мл крахмала растворяют небольшим количеством воды, затем, помешивая стеклянной палочкой, переводят в коническую колбу вместимостью 150 мл, содержащую 100 мл кипящей воды.
0,0323 н. раствор гипосульфита натрия.
Навеску 25 г кристаллического серноватистокислого (гипосульфита) натрия (NaSO·5HO), взвешенного в стеклянном стакане, переводят в мерную колбу вместимостью 1 л свежепрокипяченной охлажденной до 20 °С дистиллированной водой, добавляют 0,1 г безводного углекислого натрия (NaCO), доводят содержимое колбы до метки той же водой и оставляют в покое на 6-7 дней в темном месте. Затем из этого раствора отбирают 323 мл в мерную колбу вместимостью 1 л и доводят до метки свежепрокипяченной дистиллированной водой при 20 °С, перемешивают и устанавливают титр по 0,0323 н. раствору двухромовокислого калия (бихромата калия).
В коническую колбу вместимостью 250 мл с притертой пробкой из бюретки отмеривают точно 20 мл 0,0323 н. раствора двухромовокислого калия, прибавляют 0,5 г йодистого калия, предварительно растворенного в 5 мл воды, и 5 мл раствора серной кислоты 1:4. Колбу закрывают пробкой, смоченной раствором йодистого калия, и оставляют в темном месте в течение 10 мин. Затем прибавляют 50-60 мл воды, ополаскивая пробку водой, и титруют приготовленным раствором гипосульфита до зеленовато-желтого окрашивания, прибавляют 2 мл 0,5%-ного растворимого крахмала и продолжают титровать до перехода синей окраски в светло-зеленую. Если на 20 мл 0,0323 н. раствора двухромовокислого калия расходуется точно 20 мл приготовленного раствора гипосульфита, то последний считается точно 0,0323 н., если количество раствора гипосульфита, затраченное на титрование, отклоняется от 20 мл, то вычисляют поправочный коэффициент по формуле
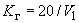 ,
,
где - количество раствора гипосульфита, пошедшее на титрование 20 мл 0,0323 н. раствора двухромовокислого калия, мл.
На этот коэффициент умножают объем гипосульфита, пошедший на титрование при определении инвертного сахара.
0,0323 н. раствор йода. 0,0323 н. раствор йода готовят из заранее приготовленного 0,1 н. раствора йода. Для этого навеску 25 г йодистого калия, взвешенного в стеклянном стакане, растворяют в 50-60 мл дистиллированной воды. В другом стакане взвешивают 12,7 г кристаллического йода и переводят в стакан с раствором йодистого калия. Полученный раствор йода в йодистом калии переводят в мерную колбу вместимостью 1 л и доводят содержимое колбы до метки свежепрокипяченной и охлажденной до 20 °С дистиллированной водой. Раствор перемешивают и переводят в стеклянную посуду с хорошо закрывающейся пришлифованной пробкой.
Мерным цилиндром отбирают 323 мл полученного раствора и переводят в мерную колбу вместимостью 1 л, содержимое колбы доводят до метки при 20 °С свежепрокипяченной дистиллированной водой, перемешивают и проверяют титр по титрованному 0,0323 н. раствору серноватистокислого (гипосульфита) натрия.
В коническую колбу вместимостью 250 мл переводят 20 мл раствора йода и титруют раствором серноватистокислого (гипосульфита) натрия от первоначального бурого до соломенно-желтого цвета. Затем прибавляют 5 мл 0,5%-ного раствора крахмала и дотитровывают до обесцвечивания. Поправочный коэффициент 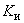 раствора йода вычисляют по формуле
раствора йода вычисляют по формуле
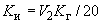 ,
,
где - количество серноватистокислого (гипосульфита) натрия, пошедшее на титрование 20 мл раствора йода, мл; 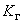 - поправочный коэффициент для раствора серноватистокислого (гипосульфита) натрия.
- поправочный коэффициент для раствора серноватистокислого (гипосульфита) натрия.
Приготовление реактива Герлеса
1,340 г азотнокислого свинца Pb(NO) растворяют в 1 л дистиллированной воды.
2,32 г гидроксида натрия растворяют в 1 л дистиллированной воды. Растворы сохраняют отдельно и для осветления мелассы расходуют в одинаковых количествах.
Раствор едкой щелочи для определения азота
330 г гидроксида натрия (NaOH) или калия (KОН) растворяют при помешивании стеклянной палочкой в фарфоровом стакане в 600 мл воды; по охлаждении раствор переливают в цилиндр и доводят объем его до 1 л.
Растворы для определения калия (в азотнокислой среде)
Реактив Тананаева для осаждения калия.
На технохимических весах отвешивают 18 г азотнокислого кобальта СО(NO)·6НО и 6 г азотнокислого серебра. Обе соли ссыпают вместе в коническую колбу вместимостью около 300 мл и растворяют в 60 мл дистиллированной воды. После растворения в ту же колбу добавляют 60 г нитрита натрия и сильно взбалтывают некоторое время. Затем прибавляют еще 50 мл воды и также взбалтывают до растворения. После этого пипеткой быстро по каплям при одновременном взбалтывании приливают 25 мл раствора HNO, содержащего 253 г безводной кислоты в объеме, доведенном дистиллированной водой до 1 л (предварительно определяют плотность исходной HNO или оттитровывают и производят расчет).
Спустя 2-3 ч (при взбалтывании через каждые 0,5 ч) раствор фильтруют через складчатый бумажный фильтр в градуированный цилиндр вместимостью 200 мл и доводят фильтрат дистиллированной водой до 195 мл. Реактив хранят в темной склянке.
0,5 н. раствор азотной кислоты. Берут 33,6 мл химически чистой кислоты относительной плотностью 1,4 и разбавляют дистиллированной водой до 1 л.
0,05%-ный водный раствор метилового оранжевого.
0,05 н. раствор азотнокислого серебра. Растворяют 8,4949 г в 1 л дистиллированной воды.
0,01 н. раствор NaOH (можно и 0,1 н.).
Промывная жидкость. 4-5 мл уксусной кислоты (80%-ной) доводят дистиллированной водой до 1 л.
Этиловый спирт
СНОН.
Серный эфир
(СН)О.
Растворы для определения содержания калия в мелассе кобальтнитритом натрия в азотнокислой среде (ускоренный метод)
20%-ный раствор кобальтнитрита натрия. Взвешивают 20 г кобальтнитрита натрия и переводят дистиллированной водой в мерную колбочку на 100 мл. После доведения до метки раствор взбалтывают и фильтруют.
1 н. раствор азотной кислоты. Берут 67,2 мл (х.ч.) кислоты относительной плотностью 1,4 и разбавляют дистиллированной водой до 1 л.
0,01 н. раствор азотной кислоты. Из имеющегося 1 н. раствора отбирают пипеткой 5 мл, вносят их в мерную колбу на 500 мл, доводят до метки дистиллированной водой и перемешивают.
Растворы для определения суммы кальциевых и магниевых солей
Необходимо иметь следующие реактивы, приготовленные с таким расчетом, чтобы 1 мл трилона Б соответствовал 1 мг СаО.
Раствор трилона Б н./28. Растворяют 6,657 г в 1 л дистиллированной воды.
Раствор хлористого кальция н./28.
1,79 г СаСО, предварительно высушенного в шкафу при 100 °С и охлажденного в эксикаторе (с хлористым кальцием), переносят в колбу вместимостью 1 л с небольшим количеством воды (до 100 мл) и добавляют сразу 2 мл, а затем по каплям концентрированной НСl до окончания выделения пузырьков СО. Когда осадок СаСО полностью растворится, колбу доливают до метки дистиллированной водой и раствор перемешивают. Этот раствор с концентрацией н./28 вместе с раствором MgSO служит для установления титра трилона Б. Магний сернокислый н./28 (4,408 г) растворяют в 1 л дистиллированной воды.
Раствор индикатора. 0,5 г хромогена-черного (специального ЕТ-00) растворяют в 100 мл аммиачно-буферного раствора и доливают этиловым спиртом до объема 100 мл.
Аммиачно-буферный раствор.
Смешивают 100 мл 20%-ного раствора NHCl (химически чистого) с 100 мл 20%-ного аммиака и смесь доливают дистиллированной водой до 1 л.
Боратно-буферный раствор.
40 г химически чистой десятиводной буры (NaBO·10НО) растворяют в 800 мл воды, прибавляют 100 мл 10%-ного гидроксида натрия. Смесь после охлаждения доводят дистиллированной водой до 1 л. Этот раствор следует хранить в склянке, плотно закрытой резиновой пробкой.
Смешанный раствор, содержащий кальций и магний в соотношении 3:1.
Раствором, составленным, например, из 300 мл н./28 СаСl и 100 мл н./28 MgSO, титруют 25 мл трилона Б, помещенных в коническую колбочку вместимостью 200-250 мл, куда добавляют также 5 мл аммиачно-буферного раствора и 7-8 капель индикатора. Титрование проводят при энергичном перемешивании до перехода окраски раствора из зеленовато-синего цвета в винно-красный.
Все реактивы, применяемые для определения солей кальция и магния, должны храниться в темном месте.
Растворы для определения магния
0,05 н. раствор трилона Б. Навеску трилона Б 9,3065 г растворяют в дистиллированной воде, фильтруют и доводят водой до 1 л.
Титр трилона Б устанавливают по металлическому цинку (по сернокислому магнию точно установить титр трудно, так как он быстро теряет кристаллизационную воду).
На аналитических весах отвешивают 1,5-2 г чистого металлического цинка, переносят в колбу на 250 мл, обрабатывают 20-30 мл соляной кислоты (1:1) под вытяжкой и слегка нагревают. После растворения цинка раствор количественно переносят в мерную колбу на 1 л, доводят до метки и перемешивают. Нормальность стандартного раствора цинка рассчитывают по формуле
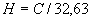 ,
,
где - навеска металлического цинка, г; 32,63 - эквивалентная масса цинка.
Поправку стандартного раствора цинка рассчитывают по формуле
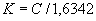 ,
,
где - навеска металлического цинка, г; 1,6342 - эквивалентная масса цинка, соответствующая 0,05 н. раствору.
Для установления нормальности раствора трилона Б отбирают пипеткой 20-25 мл стандартного раствора цинка, доливают дистиллированной водой до 100 мл, нейтрализуют несколькими каплями аммиака до избытка его по запаху, вносят 10 мл аммиачно-буферного раствора, немного сухого хромогена-черного и титруют 0,05 н. раствором трилона Б до перехода окраски индикатора из розового в синий цвет.
Нормальность трилона Б определяют по формуле
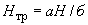 ,
,
где - нормальность стандартного раствора цинка;  - нормальность трилона Б; - объем стандартного раствора цинка, взятый на титрование, мл; - объем раствора трилона Б, пошедший на титрование, мл.
- нормальность трилона Б; - объем стандартного раствора цинка, взятый на титрование, мл; - объем раствора трилона Б, пошедший на титрование, мл.
Соляная кислота 1:1.
0,05 н. раствор сернокислого магния.
Навеску сернокислого магния MgSO·7НО в количестве 6,1625 г отвешивают на аналитических весах и растворяют в мерной колбе на 1 л дистиллированной водой.
Аммиачно-буферный раствор. 20 г хлористого аммония (химически чистого) растворяют в 100 мл дистиллированной воды, добавляют 100 мл 25%-ного раствора аммиака и разбавляют водой до 1 л.
Насыщенный раствор щавелевокислого аммония . Растворяют 5,8 г в 100 мл воды.
Растворы для определения содержания азота аминокислот "медным способом"
Раствор хлорной меди.
27,3 г CuCl·2НО переводят в колбу вместимостью 1 л и вливают дистиллированной воды до метки.
Фосфорнокислый натрий.
64,5 г NaHPO·12НО растворяют в 50 мл воды, свободной от СО, добавляют 7,2 ч NaOH, переводят в колбу вместимостью 1 л и добавляют воды до метки.
Боратный буфер.
57,21 г NaBO·10НО растворяют в 1500 мл воды, добавляют 100 мл 1 н. НСl и доводят раствор дистиллированной водой до 2000 мл.
Нормальный раствор гидроксида натрия (1 н. NaOH).
Уксусная кислота 80%-ная.
Смесь медного фосфата. Для приготовления этой смеси смешивают (по объему) одну часть раствора хлорной меди (реактив 1) с двумя частями тринатрия фосфата (реактив 2), к смеси добавляют две части боратного буфера (реактив 3).
Раствор тимолфталеина. 0,25 г тимолфталеина растворяют в 100 мл 90%-ного спирта.
Сантинормальный раствор гипосульфита.
0,01 н. NaSO·5НО 2,482 г гипосульфита растворяют в 1 л дистиллированной воды.
Раствор крахмала. Взбалтывают 1 г растворимого крахмала в 5 мл дистиллированной воды; этот раствор маленькими порциями вливают в насыщенный раствор КСl (80 мл) и кипятят 5 мин, охлаждают до комнатной температуры, переводят в колбу на 100 мл и доводят насыщенным раствором КСI до метки, раствор крахмала должен быть нейтральным (рН 7).
Приготовление нейтральной воды
К определенному количеству прокипяченной дистиллированной воды прибавляют на каждые 100 мл 2-3 капли раствора бромтимола-синего и разбавленного раствора щелочи (приблизительно 0,01 н.) до тех пор, пока вся жидкость не окрасится в голубоватый цвет.
Нейтральную воду необходимо готовить непосредственно перед использованием.
Растворы для определения хлористого калия
Алюминий хлористый ч., 0,5%-ный раствор.
Кислота уксусная х.ч., 1%-ный и 10%-ный растворы.
Тетрафенилборат натрия, 3,5%-ный раствор. Готовят следующим образом. 3,5 г тетрафенилбората натрия растворяют при комнатной температуре в 100 мл воды, приливают 0,5 мл 0,5%-ного раствора хлористого алюминия, перемешивают, после отстаивания в течение ночи в темном месте фильтруют через сухой бумажный фильтр. Первые порции фильтрата приливают обратно к фильтруемому раствору и продолжают фильтрование (применяют свежеприготовленный раствор).
Раствор промывной готовят следующим образом. К 100 мл 1%-ной уксусной кислоты прибавляют 3-4 мл 3,5%-ного раствора тетрафенилбората натрия.
Серебро азотнокислое, 1%-ный раствор.
Метиловый красный (индикатор), 0,1%-ный раствор в 60%-ном этиловом спирте.
Приготовление нейтрального раствора формалина
В колбу из белого стекла наливают 40%-ный раствор формалина, добавляют 2-3 капли индикатора фенолфталеина и осторожно из бюретки по каплям титруют крепким раствором щелочи, например 33%-ным раствором NaOH, до слабо-розового окрашивания, появляющегося от прибавления одной капли щелочи и затем исчезающего.
Формалин можно нейтрализовать и с помощью рН-метра. При этом его нейтрализуют щелочью до рН 8 (точка перехода окраски индикатора фенолфталеина).
Раствор для определения спирта методом окисления до уксусной кислоты (по Мартену)
1. 42,6070 г химически чистого, 2-3 раза перекристаллизованного и высушенного при 130 °С KСrО растворяют в 1 л воды, 1 мл этого раствора соответствует 10 мг или 0,0126 мл спирта.
2. 92 г соли Мора [FeSO(NH)SO·HO] растворяют в воде, к раствору прибавляют 20 г концентрированной серной кислоты относительной плотностью 1,84 и по охлаждении объем доводят до 1 л.
3. Свежеприготовленный раствор железосинеродистого калия 0,1 г KFe(CN) растворяют в 100 мл воды.
Приготовление шкалы эталонов для определения нитритов (окислов азота) по ХО ВНИИХП
Типовой раствор готовят, растворяя 0,1816 г химически чистого азотистокислого натрия в 1 л дистиллированной воды. 10 мл этого раствора переносят в мерную колбу вместимостью 100 мл и доливают дистиллированную воду до метки, 1 мл раствора содержит 0,00001 г NО.
Шкалу эталонов готовят следующим образом: в несколько цилиндров (одинаковых по цвету стекла и размерам) наливают по 25 мл дистиллированной воды и при помощи микробюретки добавляют в первый цилиндр 0,25 мл типового раствора, во второй - 0,5 мл и далее 0,75; 1,0; 1,5; 2,0; 2,5 мл. Затем во все цилиндры прибавляют по 5 мл реактива Грисса. Растворы в цилиндрах тщательно перемешивают и через 10-15 мин сравнивают окраску содержимого с окраской испытуемого раствора.
Реактив Грисса
Этот реактив состоит из двух растворов, хранящихся до момента использования отдельно в темных склянках. Первый раствор: 0,5 г сульфаниловой кислоты растворяют в 150 мл 30%-ной уксусной кислоты. Второй раствор: 0,1 г нафтиламина кипятят 20 мин в 20 мл воды в колбе с обратным холодильником, затем прибавляют 150 мл 30%-ной уксусной кислоты. Перед использованием оба раствора сливают вместе в равных частях.
Для приготовления реактива Грисса из сухого препарата отвешивают 1 г препарата и растворяют в 50 мл дистиллированной воды.
Приготовление бромортутной бумаги
Исходный препарат бромной ртути предварительно очищают возгонкой, для чего его помещают в фарфоровую чашку или тигель, накрывают часовым стеклом и осторожно нагревают на песчаной бане. Бромная ртуть возгоняется в виде белых игл, а в фарфоровой чашке остается черный осадок. Работа ведется в вытяжном шкафу.
Бромную ртуть можно приготовить в лаборатории: 50-100 г ртути наливают в фарфоровую чашку, обливают дистиллированной водой. Затем по каплям прибавляют к ней бром до тех пор, пока не израсходуется вся металлическая ртуть. Полученный раствор нагревают, фильтруют в горячем состоянии и дают выкристаллизоваться бромной ртути. Снова ее перекристаллизовывают из горячей воды, высушивают при возможно низкой температуре, после чего очищают возгонкой, как указано выше.
Бромортутную бумагу готовят следующим способом: полосы непроклеенной плотной белой бумаги погружают в 5%-ный спиртовой раствор бромной ртути и оставляют стоять 5-10 мин.
Необходимо следить, чтобы раствор покрывал бумагу и не было участков, не погруженных в раствор. Затем полоски вынимают, избыток раствора удаляют путем встряхивания и высушивают в горизонтальном положении на стеклянных палочках. Из высушенной бумаги вырезают полоски длиной 40-50 мм и шириной 2-2,5 мм. Готовые полоски кладут в темную склянку с хорошо притертой пробкой и хранят в темном месте (срок годности не более 3 мес).
Йодно-ртутный раствор для определения йодного числа в олеиновой кислоте
Готовят два раствора: 1) 25 г кристаллического йода в 500 мл 96%-ного этилового спирта; 2) 30 г сулемы в 500 мл 96%-ного спирта. Раствор сулемы фильтруют.
Оба раствора хранят в отдельных склянках из темного стекла с притертыми пробками и смешивают их в равных объемах за 48 ч до начала определения йодного числа.
Растворы для определения фосфора по методу Бригса
5%-ный молибденовый раствор. 25 г молибденовокислого аммония растворяют в 300 мл дистиллированной воды. К полученному раствору добавляют 75 мл концентрированной серной кислоты, предварительно разбавленной в 120 мл воды, затем общий объем раствора доводят водой до объема 500 мл.
Свежеприготовленный 1%-ный раствор гидрохинона. К 100 мл раствора добавляют одну каплю концентрированной серной кислоты для предотвращения окисления.
20%-ный раствор безводного сульфита
(NaSO). Вследствие окисления сульфитный раствор портится при стоянии, поэтому пользуются свежеприготовленным раствором.
Стандартный раствор
KНРО. Этот раствор готовят из чистой соли, которую предварительно измельчают и высушивают в эксикаторе в течение нескольких дней. 4,394 г соли растворяют дистиллированной водой в мерной колбе вместимостью 1 л, добавляют в качестве антисептика несколько капель хлороформа. 1 мл раствора содержит 1 мг фосфора. 100 мл этого раствора разбавляют водой до 1 л и получают раствор, в 1 мл которого содержится 0,1 мг фосфора.
Растворы для определения фосфора по уточненному методу Фиске и Сабуро (ХО ВНИИХП)
2,5%-ный раствор молибденовокислого аммония в 3 н. серной кислоте. На технохимических весах отвешивают в стаканчике 2,5 г молибденовокислого аммония, добавляют 5 мл дистиллированной воды, размешивают и добавляют 7,6 мл концентрированной серной кислоты, предварительно разбавленной в отдельной колбочке 30-55 мл воды. Полученный раствор молибдата аммония переводят водой в мерную колбу вместимостью 100 мл, охлаждают, доводят до метки и взбалтывают.
Восстановитель - 0,2%-ный раствор аскорбиновой кислоты. 0,2 г растворяют в 100 мл воды.
Стандартный раствор
KНРО. Этот раствор готовят из химически чистой соли, которую предварительно измельчают и высушивают на протяжении нескольких дней в эксикаторе. Взятую на аналитических весах в стаканчике навеску 2,1970 г соли растворяют дистиллированной водой и количественно переводят в мерную колбу вместимостью 500 мл. Для стойкости добавляют несколько капель хлороформа. При 20 °С раствор доводят до метки и перемешивают.
5 мл этого основного раствора пипеткой отбирают в мерную колбу на 50 мл, при 20° С доводят водой до метки и тщательно взбалтывают. В 1 мл этого раствора содержится 0,1 мг фосфора. Перед анализом каждый раз следует готовить свежий раствор.
15%-ный раствор гидроксида натрия. 75 г NaOH растворяют в 500 мл дистиллированной воды, взбалтывают и хранят в склянке с резиновой пробкой.
Растворы для определения растворенного кислорода по Винклеру
20%-ный раствор Kl. 20 г KJ растворяют в 100 мл 33%-ного раствора гидроксида натрия. Полученный раствор должен быть свободен от нитритов, при подкислении его разбавленной серной кислотой не должен окрашивать крахмал в синий цвет.
50%-ный раствор
МnСl. 50 г МnСl растворяют в 100 мл дистиллированной воды. В растворе не должно содержаться железа. Из подкисленного раствора не должен в заметных количествах выделяться йод.
0,01 н. раствор серноватистокислого натрия.
Приготовление безводной глюкозы (для определения сахара по ЛО ВНИИХП)
Безводную глюкозу получают из глюкозы марки "Медицинская" после двойной перекристаллизации из этилового 96%-ного спирта. Для этого 250-300 мл спирта-ректификата в конической колбе ставят в кипящую водяную баню. Когда спирт в колбе закипит, нагревание бани прекращают и начинают прибавлять в спирт маленькими порциями (на кончике шпателя) хорошо растертый порошок глюкозы до тех пор, пока глюкоза не перестанет растворяться.
Горячий спиртовой раствор глюкозы фильтруют через бумажный фильтр в воронке с обогревом. Фильтрат оставляют кристаллизоваться в холодном месте в течение 2 сут. Затем маточный раствор декантируют, а глюкозу с остатками спирта растирают и снова растворяют в кипящем спирте, фильтруют и для кристаллизации оставляют в холодном месте в течение 2 сут. Маточный раствор сливают, а кристаллы глюкозы растирают в фарфоровой или лучше в агатовой ступке и сушат при температуре 40-50 °С до постоянной массы в сушильном шкафу. Высушенную глюкозу хранят в склянке с хорошо притертой пробкой или в бюксе, которую помещают в эксикаторе над хлористым кальцием или над серной кислотой.
Растворы для определения сахара по методу Бертрана
1. 40 г перекристаллизованного медного купороса растворяют в воде и доводят раствор дистиллированной водой до объема 1 л.
2. 200 г сегнетовой соли (виннокислого калия-натрия) и 150 г гидроксида натрия растворяют в дистиллированной воде и раствор доводят до объема 1 л.
3. 50 г сернокислого окисного железа или 86 г железоаммиачных квасцов и 200 г крепкой серной кислоты (относительной плотностью 1,84) растворяют в дистиллированной воде и раствор доводят до объема 1 л. Прибавляя раствор KМnО, необходимо удостовериться в отсутствии закисных солей в окисной соли железа; от первой же капли должна появиться розовая окраска.
4. 5 г KМnО растворяют в 1 л дистиллированной воды. Титр раствора перманганата устанавливают по щавелевой кислоте. Для установки титра применяют предварительно перекристаллизованный щавелевокислый аммоний.
Примечание. Раствор перманганата после приготовления должен выстояться в течение 2 недель, так как перегнанная вода содержит, хотя и в небольшом количестве, вещества, способные окисляться.
Навеску щавелевокислого аммония в количестве около 0,2 г (с точностью до четвертого знака) растворяют в 50-100 мл воды, прибавляют 1-2 мл крепкой HSO, раствор нагревают до 80 °С и титруют раствором перманганата до появления розового окрашивания.
Расчет титра производят по уравнению
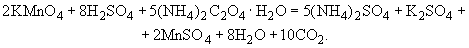
Из уравнения следует, что двум молекулам перманганата соответствует пять молекул щавелевокислого аммония, т.е. 316,08 массовой части KМnО соответствует 710,5 массовой части (NH)СО·НО.
Пример. Для определения титра перманганата взята навеска щавелевокислого аммония 0,2224 г; указанному количеству соответствует 316,08·0,2224/710,5=0,0989 г перманганата.
На титрование 0,2224 г щавелевокислого аммония израсходовано 19,78 мл раствора KМnО. Следовательно, в 20 мл содержится 0,0989 г перманганата, а 1 мл его будет соответствовать 1·0,0989/19,78=0,005 г перманганата.
В 1 л приготовленного раствора содержится 5 г KМnО. 1 мг KМnО эквивалентен 2,01 мг Сu, а 5,0 мг будут соответствовать 5·2,01=10,05 мг меди, т.е. 1 мл приготовленного раствора KМnО (титр перманганата по меди) соответствует 10,05 мг меди. На склянке, содержащей титрованный раствор KМnО, записывают количество миллиграммов меди, соответствующее 1 мл этого раствора. Раствор перманганата следует хранить в темной склянке.
Растворы для эбулиостатического метода определения сахара
Глюкоза. Для установки титра пользуются чистой безводной глюкозой, которую получают из глюкозы марки "Медицинская" после двойной перекристаллизации из этилового спирта (96%-ного). Для этого 250-300 мл спирта-ректификата наливают в коническую колбу и ставят в кипящую водяную баню. Когда спирт в колбе закипит, нагревание банки прекращают и начинают прибавлять в спирт маленькими порциями (на кончике шпателя) хорошо растертый порошок глюкозы до тех пор, пока глюкоза перестанет растворяться. Горячий спиртовой раствор глюкозы фильтруют через бумажный фильтр в воронке с обогревом. Фильтрат оставляют кристаллизоваться в холодном месте в течение 2 сут. Затем маточный раствор сливают, оставляя в колбе кристаллы глюкозы с остатками спирта. Полученные кристаллы глюкозы растирают и снова растворяют в кипящем спирте, фильтруют и оставляют в холодном месте на 2 сут для образования кристаллов. Маточный раствор сливают, а кристаллы глюкозы растирают в фарфоровой (лучше агатовой) ступке и сушат при температуре 40-50 °С в сушильном шкафу до постоянной массы.
Высушенную глюкозу хранят в склянке с хорошо притертой пробкой или в бюксе, которую помещают в эксикатор хлористым кальцием или серной кислотой.
Медный купорос.
Готовят путем многократной перекристаллизации 1 л насыщенного водного раствора CuSO при кипении, добавляют 15 мл концентрированной азотной кислоты и 20 мл горячей дистиллированной воды и снова нагревают до кипения. Горячий раствор фильтруют через бумажный фильтр в воронке с обогревом. Фильтрат собирают в кристаллизатор, в котором его быстро охлаждают, энергично перемешивая стеклянной палочкой. При этом образуются мелкие (как песок) кристаллы CuSO. Раствор с кристаллами оставляют на сутки до полного выпадания кристаллов сернокислой меди. При последней кристаллизации добиваются получения очень мелких кристаллов. После охлаждения кристаллы очень хорошо, почти досуха, отсасывают на воронке с разрежением (кристаллы должны рассыпаться), затем сушат на стеклянных пластинках при температуре не выше 30 °С (избегая освещения солнечными лучами) в течение 1-2 сут, изредка помешивая. Хранят в плотно закрытой склянке.
Раствор первый. 10 г химически чистого медного купороса и 0,04 г метиленовой сини растворяют в дистиллированной воде и объем доводят в мерной колбе до 1 л.
Раствор второй. 50 г сегнетовой соли, 75 г гидроксида натрия и 4 г желтой кровяной соли растворяют отдельно в дистиллированной воде. Растворы сливают вместе и общий объем доводят дистиллированной водой в мерной колбе до 1 л.
Установка условного титра раствора медного купороса. На аналитических весах с точностью до четвертого знака взвешивают около 0,1 г перекристаллизованной глюкозы, растворяют в дистиллированной воде и объем раствора доводят в мерной колбе до 100 мл. Точно отмеренное количество медно-щелочного раствора титруют и определяют количество миллилитров раствора глюкозы, пошедшее на титрование. Зная концентрацию раствора глюкозы и его количество, пошедшее на титрование, вычисляют количество глюкозы (в мг), расходуемое на восстановление меди, содержащейся в 5 мл раствора медного купороса или в 10 мл медно-щелочного раствора, взятого для опыта.
Пример. Приготовлен 0,09%-ный раствор глюкозы, т.е. 1 мл этого раствора содержит 0,9 мг глюкозы. На титрование 5 мл медного купороса или 10 мл медно-щелочного раствора израсходовано 6,3 мл раствора глюкозы. Следовательно, условный титр испытуемого медного раствора равен 6,3·0,9=5,67 мг.
ПРИЛОЖЕНИЕ III
ПОДГОТОВКА ПОСУДЫ, ПИТАТЕЛЬНЫХ СРЕД И КРАСОК ДЛЯ МИКРОБИОЛОГИЧЕСКОГО КОНТРОЛЯ
Посуда. Используемая посуда предварительно должна быть чисто вымыта, ополоснута дистиллированной водой, высушена и подготовлена к стерилизации.
Подготовка посуды к стерилизации. Чашки Петри завертывают в бумагу; пробирки, колбы, флаконы и другие емкости затыкают ватными пробками; пипетки - предварительно затыкают ватой верхний конец так, чтобы вата не высовывалась, завертывают в бумагу.
Стерилизация посуды. В сушильном шкафу с высоким нагревом (печь Пастера) при температуре 160-170 °С сухой жар - 1 ч. Стерилизацию посуды можно производить в автоклаве при избыточном давлении 100 кПа в течение 20-30 мин. Стерилизованную посуду после охлаждения хранят в завернутом виде в специальном отдельном шкафу.
Питательные среды. Мелассное сусло. 1-й вариант. Густую мелассу отвешивают на технических весах в количестве 200-300 г, смешивают с водой в соотношении 1:3. К смеси добавляют 0,6% объемных к массе исходной мелассы концентрированной серной кислоты и 0,4% диаммонийфосфата, тщательно размешивают и нагревают до 85 °С. Дают отстояться в высоком сосуде (в течение 2 ч). Осветленную мелассу сливают с осадка (рН раствора должен быть 5). Плотность мелассы около 20% СВ.
2-й вариант. 200-300 г густой мелассы смешивают с водой в соотношении 1:3. К раствору добавляют 0,6% объема (к массе исходной мелассы) ортофосфорной кислоты и 0,1% сернокислого аммония, тщательно перемешивают и нагревают до 85 °С. Дают отстояться в высоком сосуде в течение 2 ч. Осветленную мелассу сливают с осадка (рН раствора должен быть 5,1). Плотность около 20% СВ.
Заготовленное мелассное сусло разливают в бутылки по 350-400 мл и стерилизуют при избыточном давлении при 50 кПа 30 мин или в аппарате Коха при 100 °С 1 ч с промежутками 20-24 ч 3 сут подряд.
Для приготовления питательных сред мелассу разбавляют водой до плотности 5-8%.
Солодовое сусло. Для приготовления питательной среды можно пользоваться суслом, полученным с пивоваренного завода, неохмеленным, после фильтрации. Для приготовления сусла в лаборатории применяют солод крупного помола с высокой осахаривающей способностью. На 1 л берут 250 г солода. Смесь нагревают до 50 °С и выдерживают еще 30 мин, затем температуру осторожно повышают до 63-62,5 °С и не снижают ее до исчезновения реакции на крахмал (по йодной пробе).
Полученное сусло отделяют от дробины путем фильтрации через марлю и вату, разбавляют водой до нужной концентрации, разливают в стеклянную посуду - колбы или бутылки вместимостью 0,5 л по 300-350 мл в каждую бутылку или колбу и стерилизуют в автоклаве при избыточном давлении 50 кПа 30 мин или в аппарате Коха при 100 °С по 1 ч 3 сут подряд.
Дрожжевая вода. На 1 л водопроводной воды берут 80 г прессованных дрожжей или 20 г сушеных, смесь кипятят в течение 20-30 мин, фильтруют, разливают в бутылки на 0,5 л по 300-350 мл и стерилизуют при избыточном давлении 100 кПа в течение 1 ч. После отстаивания в течение нескольких дней дрожжевая муть оседает и дрожжевая вода становится прозрачной. Для приготовления сред прозрачную дрожжевую воду сливают с осадка.
Картофельная вода. 20 г очищенного от кожуры и протертого сырого картофеля добавляют в 1 л водопроводной воды, настаивают 4 ч, кипятят 15 мин, фильтруют через складчатый фильтр, разливают в пробирки по 5 мл и стерилизуют при избыточном давлении 100 кПа в течение 20 мин.
Дрожжевой автолизат. Для приготовления можно использовать прессованные или сушеные хлебопекарные дрожжи. 200 г прессованных дрожжей размешивают с 100 мл водопроводной воды или 100 г сушеных дрожжей с 400 мл воды, добавляют поваренной соли в количестве 0,15% к массе прессованных дрожжей и 0,5% к массе сушеных дрожжей, смесь помещают в термостат при 50 °С на 48 ч, затем нагревают до кипения и фильтруют через складчатый фильтр, разливают в стеклянную посуду и стерилизуют при избыточном давлении 100 кПа 30 мин.
Ацетатная среда для выявления посторонних дрожжевых грибков. На 1 л водопроводной воды берут 10 г уксуснокислого натрия, 10 г хлористого аммония и 5 г глюкозы. Дрожжевой автолизат добавляют в среду в количестве 3 мл. После перемешивания среду разливают в пробирки по 5 мл и стерилизуют в автоклаве при избыточном давлении 50 кПа в течение 30 мин.
Среда для поддержания производственных культур дрожжей приведена ниже.
|
Мелассное сусло концентрацией, % СВ |
10 мл |
|
Солодовое сусло концентрацией, % СВ |
90 мл |
|
Дрожжевой автолизат |
1 мл |
|
Агар (мелконарезанный) |
2 г |
Смесь доводят до кипения и кипятят на слабом огне до расплавления кусочков агара, затем фильтруют через марлю и вату, разливают в пробирки по 5 мл и стерилизуют при избыточном давлении 50 кПа в течение 20 мин или в аппарате Коха в течение 1 ч 3 сут подряд, рН среды 5,8.
Мелассный агар. К мелассному суслу, разбавленному до содержания 5-8% СВ, добавляют 2% агара и нагревают до полного расплавления кусочков его, фильтруют через марлю и вату, разливают в пробирки по 15 мл, стерилизуют при избыточном давлении 50 кПа в течение 20-30 мин или в аппарате Коха при 100 °С по 1 ч 3 сут подряд.
Сусло-агар. К солодовому суслу концентрацией 8% СВ добавляют 2% мелконарезанного агара и нагревают до кипения. После расплавления агара среду фильтруют через марлю и вату и разливают в пробирки. Стерилизуют при избыточном давлении при 50 кПа в течение 20 мин или в аппарате Коха.
Примечание. рН сусла должно быть 5,6-6. Если рН ниже 5,5, солодовое сусло подщелачивают 10%-ным раствором питьевой соды или гидроксида калия.
Сусло-желатина. К солодовому суслу концентрацией 8% СВ прибавляют 15% желатины и через 30-40 мин, когда желатина набухнет, среду подогревают до 40-50 °С, фильтруют через марлю и вату и разливают в пробирки и колбы. Стерилизуют в аппарате Коха.
Агар-дрожжевая вода с 4% сахарозы. К прозрачной, слитой с осадка дрожжевой воде добавляют 4% сахарозы и 2% мелконарезанного агара, нагревают до кипения и, когда кусочки агара расплавятся, фильтруют через марлю и вату и разливают в пробирки. Стерилизуют при 50 кПа в автоклаве в течение 20 мин или в аппарате Коха.
Среда Городковой для выявления спор у дрожжей. Количество (в г) на 100 мл водопроводной воды: пептон 1; поваренная соль 0,5; глюкоза 0,25; мелконарезанный агар 2. Среду нейтрализуют питьевой содой до рН 7,3, доводят до кипения и кипятят до тех пор, пока не расплавится агар, фильтруют через марлю и вату и разливают в пробирки по 5 мл. Стерилизуют при избыточном давлении 50 кПа в течение 20 мин.
Ацетатная среда. Состав (в %): уксуснокислый калий 0,82; дрожжевой автолизат 2,50; глюкоза 0,10; агар 2,00; водопроводная вода до 100%.
Среду нагревают до кипения и после полного расплавления агара разливают в пробирки по 5 мл. Стерилизуют при избыточном давлении 50 кПа в течение 20 мин.
Мясопептонный агар. 10 г мясного экстракта (сухого), 10 г пептона, 15-20 г агара (измельченного) смешивают в 1 л водопроводной воды и нагревают до кипения, кипятят при постоянном помешивании до полного расплавления агара. Затем смесь охлаждают до 60 °С и при помощи 1 н. раствора гидроксида натрия устанавливают рН раствора 7,7. Среду фильтруют через марлю и вату и разливают в пробирки или колбы. Стерилизуют при избыточном давлении 100 кПа в течение 20 мин. После стерилизации вторично проверяют рН (в одной из пробирок): рН среды должен остаться в пределах 7,4-7,6.
Фуксинсульфитный агар (среда Эндо). К 100 мл мясопептонного агара добавляют 1 г лактозы, 0,125 г безводного сульфита и 2,5 мл 2%-ного спиртового раствора основного фуксина в следующей последовательности. Лактозу растворяют в 2-3 мл стерильной воды при нагревании пробирки над пламенем горелки и выливают в расплавленный агар, сульфит растворяют в 3-4 мл стерильной воды в пробирке без подогревания и смешивают раствор с 2,5 мл 2%-ного спиртового раствора фуксина, смесь должна иметь заметное розовое окрашивание. После этого смесь быстро выливают в расплавленный агар, хорошо перемешивают в колбе круговым движением и разливают в несколько чашек Петри по 12-15 мл в каждую чашку.
Примечание. Чтобы заглушить рост сапрофитов, рекомендуется добавлять к фуксинсульфитному агару раствор фенола (на 100 мл среды 1 мл 5%-ного раствора кристаллического фенола).
В продаже имеется готовая сухая среда Эндо, которую готовят перед использованием согласно надписи на банке и сразу же разливают в чашки Петри.
Молочный агар. Обезжиренное молоко разливают в пробирки по 5 мл и стерилизуют дробной стерилизацией в аппарате Коха или при избыточном давлении 50 кПа в течение 20 мин.
Отдельно готовят водный агар (водопроводная вода + 3% измельченного агара). После расплавления агара разливают в пробирки по 10 мл и стерилизуют при избыточном давлении 100 кПа в течение 30 мин. Перед использованием водный агар расплавляют, соединяют с молоком и смесь выливают в чашки Петри.
Синтетическая среда с лизином. В 1 л водопроводной воды растворяют (в г): глюкозу - 50, лизин - 3, KНРО - 1, MgSO - 1, FeSO - следы. Каждый компонент растворяют в воде и добавляют в указанном порядке.
В среду вносят измельченный агар (1,5%), расплавляют и разливают в пробирки. Стерилизуют при избыточном давлении в течение 20 мин.
Среда для выявления протеолизирующих бактерий. Желатина на дрожжевой воде: 15 г желатины растворяют в 100 мл дрожжевой воды при нагревании на водяной бане. рН среды доводят до 7,2 10%-ным раствором двууглекислой соды, разливают в пробирки по 15 мл и стерилизуют при 50 кПа в течение 20 мин.
Среда с актидионом для выявления бактерий. На 1 л дрожжевой воды добавляют 20 г агара, когда он расплавится при нагревании на водяной бане, добавляют 0,1 г актидиона, доводят рН среды до 7,8-7,9 концентрированным раствором NaOH.
Среда должна быть прозрачной, если появится муть, среду осветляют белком, разливают в пробирки по 15 мл и стерилизуют при избыточном давлении 50 кПа в течение 30 мин.
Среда с лизином для выявления диких дрожжей: глюкоза - 50 г, MgSO - 1 г, KHPO - 2 г, лактат калия - 12 мл 50%-ного раствора 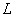 (+)-моногидрит лизина - 1 г.
(+)-моногидрит лизина - 1 г.
Витаминный раствор. На 100 мл стерильной дистиллированной воды добавляют 2 г инозитола, 0,4 г пантотената кальция, 0,5 г никотинамида, 0,1 г хлоргидрата тиамина, 20 г агара. Объем среды доводят до 1000 мл дистиллированной водой. рН среды устанавливают 5-5,2. Среду разливают в пробирки по 15 мл и стерилизуют в автоклаве в течение 15 мин при избыточном давлении 100 кПа.
Среда для выявления посторонних дрожжей, морфологически схожих с основной культурой: пептон - 10 г, KНРО - 2 г. Растворяют в 500 мл дистиллированной воды и отфильтровывают. В фильтрате расплавляют 15 г агара и добавляют 10 г глюкозы, 0,4 г эозина, 0,065 мл метиленовой сини (90%-ный спиртовой раствор). Доводят до объема 1000 мл горячей дистиллированной водой, разливают по пробиркам и стерилизуют при 100 кПа 15 мин.
При стерилизации цвет исчезает, а при охлаждении снова появляется. Хранение среды допускается не более 2 мес.
Среда для выявления сульфатредуцирующих бактерий. Основная среда: на 100 мл стерильной воды добавляют 2,3 г питательного сухого мясопептонного агара и 2 г глюкозы. Среду нагревают на водяной бане до полного растворения агара, рН среды доводят до 7,5 раствором гидроксида натрия (NaOH). Среду разливают в пробирки по 15 мл и стерилизуют 20 мин при 100 кПа.
Перед использованием к расплавленной среде добавляют по 1 мл 6,25%-ного раствора сульфита натрия и 0,4 мл 5%-ного раствора железоаммиачных квасцов.
Оба раствора перед использованием стерилизуют, пропуская через стерильный фильтр Зейтца.
Вспомогательные вещества. Стерильная водопроводная вода. Водопроводную воду разливают в пробирки по 10 или 9 мл и в колбочки по 50-100 мл, закрывают ватными пробками и стерилизуют при избыточном давлении 100 кПа в течение 30 мин или в аппарате Коха дробной стерилизацией при 100 °С по 1 ч 3 сут подряд.
Стерильный мел. Продажный мелкорастертый или размолотый мел насыпают в стерильные пробирки с ватными пробками по 0,3-0,5 г и стерилизуют сухим жаром 1 ч при 170 °С.
Раствор нистатина. Нистатин представляет собой мелкокристаллический порошок желтого цвета с активностью 4000-5000 ед. в 1 мг. Выпускаемый отечественной промышленностью антибиотик нистатин плохо растворяется в воде, поэтому при посеве используется его водная суспензия. Ее готовят так, чтобы в 1 мл суспензии содержалось количество нистатина, достаточное для подавления роста дрожжей в одной чашке Петри.
При активности нистатина 5000 ед. в мг необходимо внести в 200 мл стерильной воды навеску 200 мг. При посеве в чашки Петри вносят 1 мл этой суспензии.
При постоянной работе можно брать примерное количество нистатина без взвешивания стерильным скальпелем или химическим шпателем.
Раствор нистатина может храниться в темном холодном месте не более 5 сут. Порошок нистатина хранится без заметной потери активности в течение нескольких лет.
Порошок нистатина и его раствор нельзя подвергать стерилизации, так как при нагревании он разрушается. Однако выпускаемый нистатин обычно бывает стерильным. Для проверки стерильности при длительном хранении следует периодически высевать 1 мл его суспензии на сусло-агар или дрожжевой агар с сахарозой в чашку Петри.
Подготовка посуды и реактивов для определения биотина. Для проведения анализа необходимы следующие посуда и реактивы: 20 колбочек общей вместимостью 50 мл (конические или круглые); 5 градуированных пипеток вместимостью 5 мл; 2 пипетки Мора вместимостью 5 мл; 1 пипетка Мора вместимостью 30 мл; 10-15 градуированных пипеток на 1-2 мл; 20 пробирок, градуированных на 10 мл (одной чертой).
Обработка посуды. Вся перечисленная выше посуда должна быть тщательно вымыта и выдержана 2 ч в растворе азотной кислоты в воде (1:1). После этого посуду еще раз тщательно моют и ополаскивают дистиллированной водой, сушат, затыкают ватными пробками, а пипетки завертывают в бумагу (предварительно вводят в верхний конец комочки ваты). Посуду стерилизуют при 160-170 °С сухим паром в течение 1 ч. (Обработка азотной кислотой необходима, потому что она разрушает биотин.)
Приготовление синтетической среды. В общем объеме 1100 мл должны содержаться следующие химически чистые вещества:
|
Сахароза, г |
20 |
|
Сульфат аммония, г |
4 |
|
Фосфорнокислый натрий двузамещенный (Na |
2 |
|
Сульфат магния, г |
0,5 |
|
Хлористый кальций, г |
0,1 |
|
Железоаммиачные квасцы, г |
0,02 |
|
Сернокислый цинк, мг |
0,2 |
|
Сернокислая медь, мг |
0,01 |
|
Аневрин (В |
200 |
|
Рибофлавин (В |
100 |
|
Никотиновая кислота, мкг |
5000 |
|
Парааминобензойная кислота, мкг |
300 |
|
Пиридоксин (В |
1000 |
|
Пантотенат кальция, мкг |
500 |
|
Мезоинозитол, мкг |
50000 |
В питательную среду вводят буферный раствор, приготовленный следующим образом: 500 мл 50%-ного лактата калия + 75 мл концентрированной химически чистой молочной кислоты. Из приготовленного раствора отбирают пипеткой 10 мл и прибавляют к среде.
Чтобы упростить приготовление среды, изготовляют следующие основные растворы, которые хранят в холодильнике: раствор А, раствор В, раствор сахарозы, растворы микроэлементов, растворы витаминов.
Раствор А - 20 г сернокислого аммония (дважды перекристаллизованного) и 10 г NaHPO·2HO (дважды перекристаллизованного) растворяют в 250 мл стерильной дистиллированной воды, затем рН раствора доводят серной кислотой до 5,5
Раствор В - 2,5 г сульфата магния MgSO (перекристаллизованного), 0,5 г хлористого кальция (CaCl), 0,1 г железоаммиачных квасцов растворяют в 250 мл стерильной дистиллированной воды. Раствор В должен быть слабокислым, для этого к нему прибавляют 0,02 мл концентрированной соляной кислоты или 0,1 мл кислоты, разведенной водой в соотношении 1:4
Примечание. Железоаммиачные квасцы следует растворить отдельно в 10-15 мл воды, и, если выпадет осадок, нужно его отделить фильтрованием через складчатый фильтр и уже потом добавлять фильтрат к общему раствору.
|
Витамины |
Количество в 1 мл, мкг |
Общее количество раствора, мл |
|
|
20 |
50 |
|
|
10 |
25 |
|
Никотиновая кислота |
500 |
50 |
|
Парааминобензойная кислота |
30 |
50 |
|
Пиридоксин |
100 |
50 |
|
Пантотенат кальция |
50 |
50 |
|
Мезоинозитол |
5000 |
50 |
|
Биотин |
0,01 |
25 |
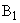
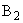
Раствор сахарозы (20%-ный) приготовляют на дистиллированной воде и для лучшей сохранности стерилизуют в аппарате Коха при 100 °С по 1 ч 3 сут подряд.
Основные растворы микроэлементов: раствор сернокислого цинка - 0,02 мг в 1 мл (0,2 мг в 10 мл дистиллированной воды); раствор сернокислой меди - 0,001 мг в 1 мл (0,01 мг в 10 мл дистиллированной воды).
Основные растворы витаминов приведены в таблице.
Все растворы готовят на стерильной дистиллированной воде в стерильной, тщательно вымытой (как описано выше) посуде и хранят в холодильнике.
Необходимая посуда для заготовки основных растворов: мерные колбы на 250 мл - 3 шт.; на 50 мл - 6-7 шт.; на 25 мл - 2-3 шт.; градуированные на 10 мл (одной чертой) пробирки - 5 шт.
Краски. Метиленовая синяя. Насыщенный спиртовой раствор: к 100 мл 96%-ного спирта (этилового) прибавляют 10 г метиленовой сини (в порошке) и оставляют стоять на несколько дней (за это время краску несколько раз встряхивают), затем раствор краски фильтруют.
Щелочная синька (по Леффлеру). В 100 мл дистиллированной воды растворяют 30 мл насыщенного спиртового раствора метиленовой сини и 1 мл 1%-ного раствора гидроксида калия.
Разбавленная синька (по Финку) для определения количества отмерших клеток. Готовят три раствора: 1) 0,009 г фосфорнокислого натрия (NaHPO) растворяют в 500 мл дистиллированной воды; 2) 13,6 г фосфорнокислого калия (KНРО) растворяют в 500 мл дистиллированной воды; 3) 0,1 г метиленовой сини (в порошке) растворяют в 500 мл дистиллированной воды.
Для приготовления рабочего раствора краски смешивают 0,25 мл первого раствора, 99,75 мл второго раствора и 100 мл третьего раствора, рН рабочего раствора должен быть 4,6.
Основной фуксин. 2 г сухой краски и 20 мл 96%-ного этилового спирта встряхивают и оставляют стоять несколько дней, получается насыщенный спиртовой раствор.
Карболовый фуксин (по Цилю) для окраски спор дрожжей. 10 мл насыщенного спиртового раствора основного фуксина прибавляют к 100 мл 5%-ного раствора карболовой кислоты.
Разбавленный фуксин (по Пфейферу). Карболовый фуксин (по Цилю) разбавляют в соотношении 1:10 дистиллированной водой - применяется при изготовлении окрашенных препаратов микроорганизмов.
Насыщенный спиртовой раствор генцианвиолет. 1 г краски (в порошке) растворяют в 10 мл 96%-ного этилового спирта и оставляют стоять несколько дней.
Карболовый раствор генцианвиолет (для окраски по Граму). Насыщенный спиртовой раствор (1 г в 10 мл 96%-ного спирта) смешивают со 100 мл 5%-ного раствора карболовой кислоты, для достижения прозрачности добавляют еще несколько капель этилового спирта.
Раствор йода в йодистом калии (по Люголю). 2 г йодистого калия растворяют в 5 мл дистиллированной воды, к полученному раствору добавляют 1 г кристаллического йода, после чего раствор доливают дистиллированной водой до 300 мл.
ПРИЛОЖЕНИЕ IV
СПРАВОЧНЫЕ ТАБЛИЦЫ
Таблица 1
Поправки для приведения показаний рефрактометра РПЛ к 20 °С
|
Температура, °С |
Концентрация сухих веществ, % |
||||||||||||||
|
0 |
5 |
10 |
15 |
20 |
25 |
30 |
35 |
40 |
45 |
50 |
55 |
60 |
65 |
70 |
|
|
Из показания рефрактометра вычесть |
|||||||||||||||
|
10 |
0,50 |
0,54 |
0,58 |
0,61 |
0,64 |
0,66 |
0,68 |
0,70 |
0,72 |
0,73 |
0,74 |
0,75 |
0,76 |
0,78 |
0,79 |
|
11 |
0,46 |
0,49 |
0,53 |
0,55 |
0,58 |
0,60 |
0,62 |
0,64 |
0,65 |
0,66 |
0,67 |
0,68 |
0,69 |
0,70 |
0,71 |
|
12 |
0,42 |
0,45 |
0,48 |
0,50 |
0,52 |
0,54 |
0,56 |
0,57 |
0,58 |
0,59 |
0,60 |
0,61 |
0,61 |
0,63 |
0,63 |
|
13 |
0,37 |
0,40 |
0,42 |
0,44 |
0,46 |
0,48 |
0,49 |
0,50 |
0,51 |
0,52 |
0,53 |
0,54 |
0,54 |
0,55 |
0,55 |
|
14 |
0,33 |
0,35 |
0,37 |
0,39 |
0,40 |
0,41 |
0,42 |
0,43 |
0,44 |
0,45 |
0,46 |
0,46 |
0,46 |
0,47 |
0,48 |
|
15 |
0,27 |
0,29 |
0,31 |
0,33 |
0,34 |
0,34 |
0,35 |
0,36 |
0,37 |
0,37 |
0,38 |
0,39 |
0,39 |
0,40 |
0,40 |
|
16 |
0,22 |
0,24 |
0,25 |
0,26 |
0,27 |
0,28 |
0,28 |
0,29 |
0,30 |
0,30 |
0,30 |
0,31 |
0,31 |
0,32 |
0,31 |
|
17 |
0,17 |
0,18 |
0,19 |
0,20 |
0,21 |
0,21 |
0,21 |
0,22 |
0,22 |
0,23 |
0,23 |
0,23 |
0,23 |
0,24 |
0,24 |
|
18 |
0,12 |
0,13 |
0,13 |
0,14 |
0,14 |
0,14 |
0,14 |
0,15 |
0,15 |
0,15 |
0,15 |
0,16 |
0,16 |
0,16 |
0,16 |
|
19 |
0,06 |
0,06 |
0,06 |
0,07 |
0,07 |
0,07 |
0,07 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
|
К показанию рефрактометра прибавить |
|||||||||||||||
|
21 |
0,06 |
0,07 |
0,07 |
0,07 |
0,07 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
0,08 |
|
22 |
0,13 |
0,13 |
0,14 |
0,14 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,16 |
0,16 |
0,16 |
0,16 |
0,16 |
0,16 |
|
23 |
0,19 |
0,20 |
0,21 |
0,22 |
0,22 |
0,23 |
0,23 |
0,23 |
0,23 |
0,24 |
0,24 |
0,24 |
0,24 |
0,24 |
0,24 |
|
24 |
0,26 |
0,27 |
0,28 |
0,29 |
0,30 |
0,30 |
0,31 |
0,31 |
0,31 |
0,31 |
0,31 |
0,32 |
0,32 |
0,32 |
0,32 |
|
25 |
0,33 |
0,35 |
0,36 |
0,37 |
0,38 |
0,38 |
0,39 |
0,40 |
0,40 |
0,40 |
0,40 |
0,40 |
0,40 |
0,40 |
0,40 |
|
26 |
0,40 |
0,42 |
0,43 |
0,44 |
0,45 |
0,46 |
0,47 |
0,48 |
0,48 |
0,48 |
0,48 |
0,48 |
0,48 |
0,48 |
0,48 |
|
27 |
0,48 |
0,50 |
0,52 |
0,53 |
0,54 |
0,55 |
0,55 |
0,56 |
0,56 |
0,56 |
0,56 |
0,56 |
0,56 |
0,56 |
0,56 |
|
28 |
0,56 |
0,57 |
0,60 |
0,61 |
0,62 |
0,63 |
0,63 |
0,64 |
0,64 |
0,64 |
0,64 |
0,64 |
0,64 |
0,64 |
0,64 |
|
29 |
0,64 |
0,66 |
0,68 |
0,69 |
0,71 |
0,72 |
0,72 |
0,73 |
0,73 |
0,73 |
0,73 |
0,73 |
0,73 |
0,73 |
0,73 |
|
30 |
0,72 |
0,74 |
0,77 |
0,78 |
0,79 |
0,80 |
0,80 |
0,81 |
0,81 |
0,81 |
0,81 |
0,81 |
0,81 |
0,81 |
0,81 |
Таблица 2
Поправки к показаниям сахарометра, калиброванного при 20 °С
|
Температура, °С |
Показания сахарометра, % |
||||||||||||
|
0 |
5 |
10 |
12 |
14 |
16 |
18 |
20 |
25 |
30 |
35 |
40 |
45 |
|
|
Из показания сахарометра вычесть |
|||||||||||||
|
0 |
0,38 |
0,47 |
0,63 |
0,69 |
0,74 |
0,78 |
0,83 |
0,87 |
0,97 |
1,06 |
1,15 |
1,24 |
1,30 |
|
5 |
0,35 |
0,45 |
0,55 |
0,59 |
0,62 |
0,66 |
0,69 |
0,72 |
0,79 |
0,85 |
0,91 |
0,97 |
1,01 |
|
10 |
0,32 |
0,37 |
0,42 |
0,44 |
0,46 |
0,48 |
0,50 |
0,51 |
0,56 |
0,60 |
0,64 |
0,67 |
0,69 |
|
11 |
0,31 |
0,34 |
0,39 |
0,41 |
0,42 |
0,44 |
0,45 |
0,47 |
0,51 |
0,54 |
0,58 |
0,61 |
0,63 |
|
12 |
0,28 |
0,31 |
0,35 |
0,37 |
0,38 |
0,40 |
0,41 |
0,42 |
0,46 |
0,49 |
0,52 |
0,54 |
0,56 |
|
13 |
0,26 |
0,28 |
0,32 |
0,33 |
0,34 |
0,35 |
0,36 |
0,38 |
0,41 |
0,43 |
0,46 |
0,48 |
0,49 |
|
14 |
0,23 |
0,25 |
0,28 |
0,29 |
0,30 |
0,31 |
0,32 |
0,33 |
0,35 |
0,37 |
0,39 |
0,41 |
0,43 |
|
15 |
0,20 |
0,21 |
0,24 |
0,25 |
0,26 |
0,26 |
0,27 |
0,28 |
0,30 |
0,31 |
0,33 |
0,35 |
0,36 |
|
16 |
0,16 |
0,18 |
0,19 |
0,20 |
0,21 |
0,22 |
0,22 |
0,23 |
0,23 |
0,25 |
0,27 |
0,28 |
0,29 |
|
17 |
0,12 |
0,13 |
0,15 |
0,15 |
0,16 |
0,16 |
0,17 |
0,17 |
0,18 |
0,19 |
0,20 |
0,21 |
0,22 |
|
18 |
0,08 |
0,09 |
0,10 |
0,10 |
0,11 |
0,11 |
0,11 |
0,12 |
0,12 |
0,13 |
0,13 |
0,14 |
0,15 |
|
19 |
0,04 |
0,05 |
0,05 |
0,05 |
0,05 |
0,05 |
0,06 |
0,06 |
0,06 |
0,07 |
0,07 |
0,07 |
0,08 |
|
20 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
|
К показанию сахарометра прибавить |
|||||||||||||
|
21 |
0,05 |
0,05 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,07 |
0,07 |
0,07 |
0,07 |
|
22 |
0,10 |
0,11 |
0,11 |
0,11 |
0,12 |
0,12 |
0,12 |
0,12 |
0,13 |
0,14 |
0,14 |
0,14 |
0,15 |
|
23 |
0,15 |
0,16 |
0,17 |
0,17 |
0,18 |
0,18 |
0,18 |
0,18 |
0,20 |
0,21 |
0,22 |
0,22 |
0,22 |
|
24 |
0,21 |
0,22 |
0,23 |
0,24 |
0,24 |
0,25 |
0,25 |
0,26 |
0,26 |
0,28 |
0,29 |
0,30 |
0,30 |
|
25 |
0,27 |
0,28 |
0,30 |
0,30 |
0,31 |
0,31 |
0,32 |
0,32 |
0,33 |
0,35 |
0,36 |
0,37 |
0,38 |
|
26 |
0,32 |
0,34 |
0,36 |
0,37 |
0,37 |
0,38 |
0,39 |
0,39 |
0,41 |
0,42 |
0,44 |
0,45 |
0,45 |
|
27 |
0,40 |
0,41 |
0,42 |
0,43 |
0,44 |
0,45 |
0,46 |
0,46 |
0,48 |
0,50 |
0,51 |
0,53 |
0,53 |
|
28 |
0,46 |
0,47 |
0,49 |
0,50 |
0,51 |
0,52 |
0,53 |
0,54 |
0,55 |
0,58 |
0,59 |
0,61 |
0,61 |
|
29 |
0,53 |
0,54 |
0,56 |
0,57 |
0,58 |
0,59 |
0,60 |
0,61 |
0,63 |
0,65 |
0,67 |
0,69 |
0,70 |
|
30 |
0,60 |
0,61 |
0,63 |
0,64 |
0,65 |
0,66 |
0,67 |
0,68 |
0,71 |
0,73 |
0,75 |
0,77 |
0,78 |
|
35 |
0,99 |
0,99 |
1,02 |
1,03 |
1,05 |
1,06 |
1,08 |
1,09 |
1,13 |
1,16 |
1,17 |
1,19 |
1,20 |
|
40 |
1,41 |
1,43 |
1,46 |
1,48 |
1,50 |
1,51 |
1,53 |
1,54 |
1,57 |
1,60 |
1,61 |
1,63 |
1,64 |
|
50 |
2,46 |
2,47 |
2,50 |
2,51 |
2,52 |
2,53 |
2,54 |
2,55 |
2,57 |
2,58 |
2,58 |
2,58 |
2,58 |
Таблица 3
Содержание этилового спирта в культуральной среде при выращивании хлебопекарных дрожжей
|
|
Содержание спирта (в %) при расходе гипосульфита в глухом опыте, мл |
||||||||||
|
8,0 |
8,1 |
8,2 |
8,3 |
8,4 |
8,5 |
8,6 |
8,7 |
8,8 |
8,9 |
9,0 |
|
|
0,1 |
0,02 |
0,02 |
0,02 |
0,02 |
0,01 |
0,01 |
0,01 |
0,01 |
0,01 |
0,01 |
0,01 |
|
0,2 |
0,03 |
0,03 |
0,03 |
0,03 |
0,03 |
0,03 |
0,03 |
0,03 |
0,03 |
0,03 |
003 |
|
0,3 |
0,05 |
0,05 |
0,05 |
0,05 |
0,04 |
0,04 |
0,04 |
004 |
0,04 |
0,04 |
0,04 |
|
0,4 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
0,06 |
|
0,5 |
0,08 |
0,08 |
0,08 |
0,08 |
0,07 |
0,07 |
0,07 |
0,07 |
0,07 |
0,07 |
0,07 |
|
0,6 |
0,09 |
0,09 |
0,09 |
0,09 |
0,09 |
0,09 |
0,09 |
0,09 |
0,09 |
0,08 |
0,08 |
|
0,7 |
0,11 |
0,11 |
0,11 |
0,11 |
0,10 |
0,10 |
0,10 |
0,10 |
0,10 |
0,10 |
0,10 |
|
0,8 |
0,13 |
0,12 |
0,12 |
0,12 |
0,12 |
0,12 |
0,12 |
0,11 |
0,11 |
0,11 |
0,11 |
|
0,9 |
0,14 |
0,14 |
0,14 |
0,14 |
0,13 |
0,13 |
0,13 |
0,13 |
0,13 |
0,13 |
0,13 |
|
1,0 |
0,16 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,14 |
0,14 |
0,14 |
0,14 |
|
1,1 |
0,17 |
0,17 |
0,17 |
0,17 |
0,16 |
0,16 |
0,16 |
0,16 |
0,16 |
0,15 |
0,15 |
|
1,2 |
0,19 |
0,18 |
0,18 |
0,18 |
0,18 |
0,17 |
0,17 |
0,17 |
0,17 |
0,17 |
0,17 |
|
1,3 |
0,20 |
0,20 |
0,20 |
0,20 |
0,19 |
0,19 |
0,19 |
0,19 |
0,19 |
0,18 |
0,18 |
|
1,4 |
0,22 |
0,22 |
0,21 |
0,21 |
0,21 |
0,20 |
0,20 |
0,20 |
0,20 |
0,20 |
0,19 |
|
1,5 |
0,23 |
0,23 |
0,23 |
0,23 |
0,22 |
0,22 |
0,22 |
0,22 |
0,21 |
0,21 |
0,21 |
|
1,6 |
0,25 |
0,25 |
0,24 |
0,24 |
0,24 |
0,24 |
0,23 |
0,23 |
0,23 |
0,23 |
0,22 |
|
1,7 |
0,27 |
0,26 |
0,26 |
0,26 |
0,25 |
0,25 |
0,25 |
0,24 |
0,24 |
0,24 |
0,24 |
|
1,8 |
0,28 |
0,28 |
0,27 |
0,27 |
0,27 |
0,27 |
0,26 |
0,26 |
0,26 |
0,25 |
0,25 |
|
1,9 |
0,30 |
0,29 |
0,29 |
0,29 |
0,28 |
0,28 |
0,28 |
0,27 |
0,27 |
0,27 |
0,26 |
|
2,0 |
0,31 |
0,31 |
0,30 |
0,30 |
0,30 |
0,29 |
0,29 |
0,29 |
0,28 |
0,28 |
0,28 |
|
2,2 |
0,34 |
0,34 |
0,34 |
0,33 |
0,33 |
0,32 |
0,32 |
0,32 |
0,31 |
0,31 |
0,31 |
|
2,3 |
0,38 |
0,37 |
0,37 |
0,36 |
0,36 |
0,35 |
0,35 |
0,34 |
0,34 |
0,34 |
0,33 |
|
2,5 |
0,39 |
0,39 |
0,38 |
0,38 |
0,37 |
0,37 |
0,36 |
0,36 |
0,35 |
0,35 |
0,35 |
|
2,6 |
0,41 |
0,41 |
0,39 |
0,39 |
0,38 |
0,38 |
0,38 |
0,37 |
0,37 |
0,37 |
0,36 |
|
2,8 |
0,44 |
0,43 |
0,43 |
0,42 |
0,42 |
0,41 |
0,41 |
0,40 |
0,40 |
0,39 |
0,39 |
|
3,0 |
0,47 |
0,46 |
0,45 |
0,45 |
0,45 |
0,44 |
0,44 |
0,43 |
0,43 |
0,42 |
0,42 |
|
3,5 |
0,55 |
0,54 |
0,53 |
0,52 |
0,52 |
0,52 |
0,51 |
0,50 |
0,50 |
0,49 |
0,49 |
|
4,0 |
0,63 |
0,62 |
0,61 |
0,60 |
0,60 |
0,59 |
0,58 |
0,58 |
0,57 |
0,56 |
0,56 |
|
4,2 |
0,66 |
0,65 |
0,64 |
0,63 |
0,63 |
0,62 |
0,61 |
0,60 |
0,60 |
0,59 |
0,58 |
|
4,4 |
0,69 |
0,68 |
0,67 |
0,66 |
0,66 |
0,65 |
0,64 |
0,63 |
0,62 |
0,62 |
0,61 |
|
4,5 |
0,70 |
0,69 |
0,69 |
0,68 |
0,67 |
0,66 |
0,65 |
0,65 |
0,64 |
0,63 |
0,63 |
|
4,6 |
0,72 |
0,71 |
0,70 |
0,69 |
0,68 |
0,68 |
0,67 |
0,66 |
0,65 |
0,65 |
0,64 |
|
4,8 |
0,75 |
0,74 |
0,73 |
0,72 |
0,71 |
0,71 |
0,70 |
0,69 |
0,68 |
0,66 |
0,66 |
|
5,0 |
0,78 |
0,77 |
0,76 |
0,75 |
0,74 |
0,74 |
0,73 |
0,72 |
0,71 |
0,70 |
0,69 |
|
5,2 |
0,81 |
0,80 |
0,79 |
0,78 |
0,77 |
0,77 |
0,76 |
0,75 |
0,74 |
0,73 |
0,72 |
|
5,4 |
0,84 |
0,83 |
0,82 |
0,81 |
0,80 |
0,79 |
0,78 |
0,78 |
0,77 |
0,76 |
0,75 |
|
5,5 |
0,86 |
0,85 |
0,83 |
0,83 |
0,82 |
0,81 |
0,80 |
0,79 |
0,78 |
0,77 |
0,76 |
|
5,6 |
0,87 |
0,86 |
0,85 |
0,84 |
0,83 |
0,82 |
0,81 |
0,81 |
0,80 |
0,79 |
0,78 |
|
5,8 |
0,91 |
0,90 |
0,88 |
0,87 |
0,86 |
0,85 |
0,84 |
0,83 |
0,82 |
0,82 |
0,82 |
|
6,0 |
0,94 |
0,93 |
0,92 |
0,90 |
0,89 |
0,88 |
0,87 |
0,86 |
0,85 |
0,84 |
0,83 |
|
6,2 |
0,97 |
0,96 |
0,95 |
0,93 |
0,92 |
0,91 |
0,90 |
0,89 |
0,88 |
0,87 |
0,86 |
|
6,4 |
1,00 |
0,99 |
0,98 |
0,96 |
0,95 |
0,94 |
0,93 |
0,92 |
0,91 |
0,90 |
0,89 |
|
6,5 |
1,02 |
1,00 |
0,99 |
0,98 |
0,97 |
0,96 |
0,94 |
0,93 |
0,92 |
0,91 |
0,90 |
|
6,6 |
1,03 |
1,02 |
1,01 |
0,99 |
0,98 |
0,97 |
0,96 |
0,95 |
0,94 |
0,93 |
0,92 |
|
6,8 |
1,06 |
1,05 |
1,04 |
1,02 |
1,01 |
1,00 |
0,99 |
0,98 |
0,97 |
0,96 |
0,95 |
|
7,0 |
1,09 |
1,08 |
1,07 |
1,05 |
1,04 |
1,03 |
1,02 |
1,02 |
1,01 |
0,99 |
0,97 |
|
7,5 |
1,18 |
1,16 |
1,14 |
1,13 |
1,12 |
1,10 |
1,09 |
1,08 |
1,07 |
1,05 |
1,04 |
 , мл
, млТаблица 4
Определение концентрации дрожжей в дрожжевом молоке и количества дрожжевого молока для определения в нем качества дрожжей
|
Содержание СВ, % |
Концентрация дрожжей |
Количество дрожжевого молока (в мл) для определения |
||
|
подъемной силы дрожжей |
кислотности |
|||
|
арбитражным методом |
методом шарика |
|||
|
8,0 |
407 |
12 |
7,7 |
25 |
|
8,1 |
413 |
12 |
7,7 |
24 |
|
8,2 |
419 |
12 |
7,4 |
24 |
|
8,3 |
426 |
12 |
7,3 |
23 |
|
8,4 |
432 |
12 |
7,2 |
23 |
|
8,5 |
436 |
12 |
7,1 |
23 |
|
8,6 |
440 |
11 |
7,1 |
23 |
|
8,7 |
444 |
11 |
7,0 |
22 |
|
8,8 |
449 |
11 |
7,0 |
22 |
|
8,9 |
453 |
11 |
6,9 |
22 |
|
9,0 |
457 |
11 |
6,8 |
22 |
|
9,1 |
465 |
11 |
6,7 |
21 |
|
9,2 |
474 |
11 |
6,6 |
21 |
|
9,3 |
482 |
10 |
6,5 |
21 |
|
9,4 |
490 |
10 |
6,4 |
20 |
|
9,5 |
498 |
10 |
6,3 |
20 |
|
9,6 |
507 |
10 |
6,2 |
20 |
|
9,7 |
511 |
10 |
6,1 |
20 |
|
9,8 |
514 |
10 |
6,1 |
19 |
|
9,9 |
518 |
10 |
6,1 |
19 |
|
10,0 |
521 |
10 |
6,0 |
19 |
|
10,1 |
525 |
10 |
6,0 |
19 |
|
10,2 |
528 |
9 |
5,9 |
19 |
|
10,3 |
532 |
9 |
5,9 |
19 |
|
10,4 |
538 |
9 |
5,8 |
18 |
|
10,5 |
544 |
9 |
5,7 |
18 |
|
10,6 |
551 |
9 |
5,7 |
18 |
|
10,7 |
557 |
9 |
5,6 |
18 |
|
10,8 |
562 |
9 |
5,6 |
18 |
|
10,9 |
557 |
9 |
5,6 |
18 |
|
11,0 |
572 |
9 |
5,5 |
17 |
|
11,1 |
577 |
9 |
5,4 |
17 |
|
11,2 |
582 |
9 |
5,4 |
17 |
|
11,3 |
586 |
9 |
5,3 |
17 |
|
11,4 |
592 |
9 |
5,3 |
17 |
|
11,5 |
598 |
9 |
5,2 |
17 |
|
11,6 |
603 |
8 |
5,2 |
16 |
|
11,7 |
608 |
8 |
5,1 |
16 |
|
11,8 |
613 |
8 |
5,1 |
16 |
|
11,9 |
618 |
8 |
5,1 |
16 |
|
12,0 |
623 |
8 |
5,0 |
16 |
|
12,1 |
628 |
8 |
5,0 |
16 |
|
12,2 |
633 |
8 |
4,9 |
16 |
|
12,3 |
638 |
8 |
4,9 |
16 |
|
12,4 |
643 |
8 |
4,9 |
16 |
|
12,5 |
648 |
8 |
4,8 |
15 |
|
12,6 |
653 |
8 |
4,8 |
15 |
|
12,7 |
658 |
8 |
4,8 |
15 |
|
12,8 |
663 |
8 |
4,7 |
15 |
|
12,9 |
668 |
8 |
4,7 |
15 |
|
13,0 |
674 |
7 |
4,6 |
15 |
|
13,1 |
679 |
7 |
4,6 |
15 |
|
13,2 |
684 |
7 |
4,6 |
15 |
|
13,3 |
689 |
7 |
4,5 |
15 |
|
13,4 |
694 |
7 |
4,5 |
14 |
|
13,5 |
699 |
7 |
4,5 |
14 |
|
13,6 |
704 |
7 |
4,4 |
14 |
|
13,7 |
709 |
7 |
4,4 |
14 |
Таблица 5
Соотношение между количеством меди и сахара (в мг) при определении по методу Бертрана
|
Сахар, мг |
Медь |
|
10 |
20,4 |
|
11 |
22,4 |
|
12 |
24,3 |
|
13 |
26,3 |
|
14 |
28,3 |
|
15 |
30,2 |
|
16 |
32,2 |
|
17 |
34,2 |
|
18 |
36,2 |
|
19 |
38,1 |
|
20 |
40,1 |
|
21 |
42,0 |
|
22 |
43,9 |
|
23 |
45,8 |
|
24 |
47,7 |
|
25 |
49,6 |
|
26 |
51,5 |
|
27 |
53,4 |
|
28 |
55,3 |
|
29 |
57,2 |
|
30 |
59,1 |
|
31 |
60,9 |
|
32 |
62,8 |
|
33 |
64,6 |
|
34 |
66,5 |
|
35 |
68,3 |
|
36 |
70,1 |
|
37 |
72,0 |
|
38 |
73,8 |
|
39 |
75,7 |
|
40 |
77,5 |
|
41 |
79,3 |
|
42 |
81,1 |
|
43 |
82,9 |
|
44 |
84,7 |
|
45 |
86,4 |
|
46 |
88,2 |
|
47 |
90,0 |
|
48 |
91,8 |
|
49 |
93,6 |
|
50 |
95,4 |
|
51 |
97,1 |
|
52 |
98,9 |
|
53 |
100,3 |
|
54 |
102,3 |
|
55 |
104,1 |
|
56 |
105,8 |
|
57 |
107,6 |
|
58 |
109,3 |
|
59 |
111,1 |
|
60 |
112,8 |
|
61 |
114,5 |
|
62 |
116,2 |
|
63 |
117,9 |
|
64 |
119,6 |
|
65 |
121,3 |
|
66 |
123,0 |
|
67 |
124,7 |
|
68 |
126,4 |
|
69 |
128,1 |
|
70 |
129,8 |
|
71 |
131,4 |
|
72 |
133,1 |
|
73 |
134,7 |
|
74 |
136,3 |
|
75 |
137,9 |
|
76 |
139,6 |
|
77 |
141,2 |
|
78 |
142,8 |
|
79 |
144,5 |
|
80 |
146,1 |
|
81 |
147,7 |
|
82 |
149,3 |
|
83 |
150,9 |
|
84 |
152,5 |
|
85 |
154,0 |
|
86 |
155,6 |
|
87 |
157,2 |
|
88 |
158,8 |
|
89 |
160,4 |
|
90 |
162,0 |
|
91 |
163,6 |
|
92 |
165,2 |
|
93 |
166,7 |
|
94 |
168,3 |
|
95 |
169,9 |
|
96 |
171,5 |
|
97 |
173,1 |
|
98 |
174,6 |
|
99 |
176,2 |
|
100 |
177,8 |
ПРИЛОЖЕНИЕ V
МЕТРОЛОГИЧЕСКАЯ КАРТА КОНТРОЛЯ ПРОИЗВОДСТВЕННЫХ ПРОЦЕССОВ
|
Контролируемый параметр |
Средство измерения |
Примечание |
|||||
|
Наименование, единица измерения |
Область значения |
Допус- |
Наименование, тип |
Шкала, диапазон |
Основная погреш- |
||
|
Сырье и материалы |
|||||||
|
I. Меласса |
|||||||
|
Концентрация сухих веществ, % СВ |
74-85 |
±1,0 |
Сахарометр типа Д или Г |
- |
0,1 |
Шкала в зависимости от разбавления |
|
|
Рефрактометр РПЛ-3 и др. |
0-95% СВ |
0,2 |
|||||
|
рН |
6,5-8,5 |
±0,2 |
Лабораторный рН-метр (рН-340, РН-673 и др.) |
0-14 |
±0,05 |
||
|
Автоматический рН-метр (рН-261,рН-102 и др.) |
3-8 |
±0,1 |
|||||
|
Содержанке сахара, определяемое поляриметром, % |
40,5-50,0 |
±0,2 |
Поляриметр СУ-3 и др. |
-40 |
±0,1 °S |
1 °S=1% сахара при навеске мелассы согласно инструкции |
|
|
Плотность, г/см |
1,372- |
±0,001 |
Денсиметр типа IIа |
- |
±0,001 |
- |
|
|
Основные материалы |
|||||||
|
II. Экстракт кукурузный сгущенный |
|||||||
|
Концентрация сухих веществ, % СВ |
48-60 |
±1,0 |
Весы аналитические ВЛР-200 г и др. |
0-200 |
±0,0001 |
Высушивание навески 2-3 г |
|
|
Вспомогательные материалы |
|||||||
|
III. Кислота серная техническая |
|||||||
|
Плотность, г/см |
1,831- |
0,001 |
Денсиметр типа IIа |
- |
0,001 |
||
|
IV. Кислота олеиновая техническая марок А и Б |
|||||||
|
Температура застывания, °С |
0-10 |
±0,5 |
Термометр ртутный |
0-50 |
±0,5 |
||
|
Содержание влаги, % |
0,1-0,5 |
±1,0 |
определяют по ГОСТу |
||||
|
±0,1 |
|||||||
|
Подготовительное отделение |
|||||||
|
Концентрация сухих веществ, % СВ |
10-45 |
±0,5 |
Сахарометр типа Д |
10-20, |
±0,1 |
Для раствора солей |
|
|
Автоматический рефрактометр ЕДР |
0-95 |
±0,2 |
|||||
|
Лабораторный рефрактометр РПЛ-3 |
0-25, |
±0,5 |
|||||
|
рН |
4-6 |
±0,2 |
Лабораторный рН-метр (рН-340, рН-673 и др.) |
0-14 |
±0,05 |
||
|
Автоматический рН-метр (рН-261, рН-102 и др.) |
3-8 |
±0,1 |
|||||
|
Содержание сахара, определяемое поляриметром, % |
18-30 |
±0,2 |
Поляриметр СУ-3 |
-40 |
±0,1 °S |
||
|
Температура нагрева мелассы, °С |
80-110 |
±1,0 |
Ртутный термометр |
0-150 |
±1,0 |
||
|
Температура мелассы при подаче в дрожжерастильный аппарат, °С |
10-30 |
±1,0 |
То же |
0-50 |
±0,5 |
||
|
Отделение чистой культуры (ЧК) |
|||||||
|
Температура мелассы, °С |
10-30 |
±1,0 |
" |
0-50 |
±0,5 |
В зависимости от применяемой стадии и схемы выращивания |
|
|
Концентрация сухих веществ, % СВ |
3-13 |
±0,2 |
Сахарометр типа Д |
0-10 |
±0,1 |
То же |
|
|
10-20 |
±0,1 |
||||||
|
рН |
4,5-5,2 |
±0,2 |
Лабораторный рН-метр |
0-14 |
±0,5 |
" |
|
|
Температура среды, °С |
24-31 |
±1,0 |
Ртутный термометр |
0-50 |
±0,5 |
" |
|
|
Набор среды в аппарате, м |
0,22-75 |
±1,0 |
Водомерное стекло и др. |
- |
- |
По тарировочным таблицам |
|
|
Температура воздуха, °С |
28-35 |
±1,0 |
Термометр ртутный |
0-50 |
±0,5 |
||
|
Количество воздуха, поступающего в аппараты, м |
0,2-6000 |
±1,5 |
Типовой комплект расходомера |
0-5 |
±1,5% |
В зависимости от применяемой схемы и стадии выращивания |
|
|
Концентрация дрожжей, г/л |
17-140 |
±1,5 |
Весы ВЛКТ-500М и др. |
0-500 г |
±20 мг |
То же |
|
|
Формольное число, мл 0,1 н. раствора щелочи |
0,6-7,0 |
±0,1 |
Бюретка |
0-25 мл |
±0,1 |
" |
|
|
Отделение естественно-чистой культуры (ЕЧК) |
|||||||
|
Температура мелассы, °С |
10-30 |
±1,0 |
Ртутный термометр |
0-50 |
±0,5 |
" |
|
|
Концентрация сухих веществ, % СВ |
3,0-12,0 |
±0,2 |
Сахарометр типа Д |
0-10 |
±0,1 |
" |
|
|
10-15 |
±0,1 |
" |
|||||
|
рН |
4,5-5,5 |
±0,2 |
Лабораторный рН-метр |
0-14 |
±0,5 |
" |
|
|
Температура среды, °С |
26-32 |
±1,0 |
Ртутный термометр |
0-50 |
±0,5 |
" |
|
|
Набор среды в аппарате, м |
5,6-33 |
±1,0 |
Водомерное стекло и др. |
- |
- |
По тарировочным таблицам |
|
|
Температура воздуха, °С |
28-35 |
±1,0 |
Ртутный термометр |
0-50 |
±0,5 |
||
|
Количество воздуха, поступающего в аппараты, м |
200-3000 |
±1,5 |
Типовой комплект расходомера |
0-5 |
±1,5% |
В зависимости от применяемой схемы и стадии выращивания |
|
|
Концентрация дрожжей, г/л |
8,0-61,0 |
+1,5 |
Весы ВЛКТ-500М и др. |
0-500 г |
±20 мг |
В зависимости от применяемой схемы и стадии выращивания |
|
|
Формольное число, мл 0,1 н. раствора щелочи |
1,5-3,5 |
±0,1 |
Бюретка |
0-25 мл |
±0,1 |
То же |
|
|
Товарное дрожжерастильное отделение |
|||||||
|
Температура, °С |
24-33 |
±1,5 |
Термометр СП-2, лагометр ЛР-64-02 |
0-50 |
±1,5 |
" |
|
|
0-100 |
±1,5 |
||||||
|
Концентрация сухих веществ, % СВ |
1,2-8,5 |
±0,2 |
Сахарометр типа Д |
0-10 |
±0,1 |
" |
|
|
рН |
4,5-5,8 |
±0,2 |
Лабораторный рН-метр |
0-14 |
±0,05 |
" |
|
|
Автоматический рН-метр (рН-261, рН-102 и др.) |
3-8 |
±0,1 |
|||||
|
Формольное число, мл 0,1 н. раствора щелочи |
0,1-2,0 |
±0,1 |
Бюретка |
0-25 мл |
±0,1 |
" |
|
|
Набор среды в аппарате, м |
4,6-140 |
±1,0 |
Манометр ОБМ-160 |
0-1 кгс/см |
±1,5 |
" |
|
|
±3,0 |
|||||||
|
Концентрация дрожжей, г/л |
40-140 |
±1,5 |
Весы ВЛКТ-500М и др. |
0-500 г |
±20 мг |
" |
|
|
Температура воздуха, °С |
20-60 |
±1,0 |
Ртутный термометр |
0-100 |
±1,0 |
||
|
Количество воздуха, поступающего в аппарат, м |
500-4200 |
±1,5 |
Типовой комплект для измерения расхода |
0,5-12,5 |
±1,5 |
" |
|
|
Выделение товарных дрожжей |
|||||||
|
Температура, °С |
2-12 |
±1,0 |
Термометр СП-2 |
0-50 |
±1,5% |
||
|
Лагометр ЛР-64 |
|||||||
|
Концентрация дрожжей г/л |
450-800 |
±1,0 |
Весы ВЛКТ-500М |
0-500 г |
±20 мг |
||
|
Сахарометр типа Е |
|||||||
|
Упаковка готовой продукции |
|||||||
|
Влажность дрожжей, % |
67-75 |
±0,05 |
Весы ВЛР-200 г |
0-200 г |
±0,0001 |
||
|
Масса бруска, г |
50-1000 |
Весы технические ВНЦ-2, ВНЦ-10 и др. |
0-200 |
±1% |
В зависимости от массы бруска |
||
|
0-1000 |
±1% |
||||||
|
Холодильная камера |
|||||||
|
Температура, °С |
0-4 |
±1,0 |
Ртутный термометр ТБ-2М |
0-35 |
±1,0 |
||
|
Лагометр ЛР-64 |
0-50 |
±1,5% |
|||||
|
Относительная влажность воздуха, % |
82-96 |
±5 |
Психрометр ПБ-1А и др. |
0-100 |
±0,5 °С |
||
|
Отделение сушки |
|||||||
|
Температура, °С |
|||||||
|
поступающего воздуха |
30-80 |
±1,5 |
Комплект приборов к сушильному агрегату |
- |
- |
||
|
отходящего воздуха |
25-35 |
±1,5 |
То же |
- |
- |
||
|
Масса единичной фасованной продукции, г |
10-100 100-2000 |
±3,0 ±1,0 |
Весы BHЦ-2 и др. Весы ВЛТ |
|
0-200 |
±1,0% |
|
|
Масса нефасованной продукции, кг |
10-25 |
±0,5 |
Весы ДДР-50 |
15-25 |
±0,5% |
||
|
Весы РП-200Ш13 |
10-200 |
±50 г |
В диапазоне 10-50 кг |
||||
|
Склад сушеных дрожжей |
|||||||
|
Температура воздуха, °С |
0-15 |
±1,0 |
Ртутный термометр ТБ-2М |
0-35 |
±1,0 |
||
|
Лагометр ЛР-64 и др. |
0-50 |
±1,5% |
|||||
|
Относительная влажность воздуха, % |
35-100 |
±0,5 |
Психрометр ПБ-1А и др. |
0-100 |
±0,5 °С |
||
|
Дрожжевое молоко |
|||||||
|
Концентрация сухих веществ, % СВ |
6,5-13,9 |
±0,2 |
Сахарометр типа Е |
0-8, 8-16 |
±0,05 |
||
|
Концентрация дрожжей, г/л |
400-720 |
±1,5 |
Весы ВЛКТ-500М и др. |
0-500 г |
±20 мг |
||
|
Температура, °С |
+2-+15 |
±1,0 |
Ртутный термометр |
0-50 |
±0,5 |
||
|
Количество дрожжевого молока |
0,22-20 |
±1,0 |
Водомерное стекло и др. |
- |
- |
По тарировочным таблицам |
|
|
рН |
5,5-5,2 |
±0,2 |
Лабораторный рН-метр (рН-340, рН-673 и др.) |
0-14 |
±0,05 |
||
|
Влажность отфильтрованного пласта дрожжей, % |
67-75 |
±0,05 |
Весы ВЛР-200 |
0-200 |
±0,0001 |
||
|
Масса дрожжей, г |
50-200 |
±1,5 |
Весы технические ВЛКТ-500М и др. |
0-500 |
±20 мг |
||
Представленные в схеме химико-бактериологического контроля (см. приложение VII*) показатели, обозначенные римскими цифрами, прилагаются для всех дрожжевых предприятий независимо от мощности. На заводах производительностью более 6000 т дрожжей в год рекомендуется вести контроль производства по полной схеме (показатели обозначены римскими и арабскими цифрами).
_______________
* Нумерация соответствует оригиналу. - Примечание изготовителя базы данных.
СХЕМА ХИМИКО-БАКТЕРИОЛОГИЧЕСКОГО КОНТРОЛЯ ДРОЖЖЕВОГО ПРОИЗВОДСТВА
|
Объект лабораторного контроля |
Место контроля |
Контролируемые параметры |
Периодичность контроля |
Примечание |
|||||||||
|
Сырье |
|||||||||||||
|
Меласса при отборе |
Сахарные заводы |
I. Сухие вещества, % II. рН III. Сахар по поляриметру, % IV. Доброкачественность, % V. Инвертный сахар, % VI. Сумма сбраживаемых сахаров, % |
Из каждой отобранной пробы |
||||||||||
|
Меласса при приеме |
Цистерны |
I. Сухие вещества, % II. рН III. Сахар по поляриметру, % IV. Инвертный сахар, % V. Раффиноза, % VI. Сумма сбраживаемых сахаров, % VII. Доброкачественность, % VIII. Азот, % IX. Нитриты по биопробе |
При получении каждой партии |
Партия мелассы - это количество ее, одновременно поступившее с одного сахарного завода. В случае приемки в течение 1 сут нескольких цистерн с мелассой с одного и того же завода допускается составление одной средней пробы из нескольких цистерн, но не более чем из 10 |
|||||||||
|
1. Зола, % ("условная углекислая" и "за вычетом углекислого кальция") |
|||||||||||||
|
2. Цветность, мл 0,1 н. раствора йода на 100 мл 2%-ного раствора мелассы |
|||||||||||||
|
3. Калий (K 4. Магний (MgO), % 5. Кальций (СаО), % |
|
Анализ проводится по мере надобности |
|||||||||||
|
Меласса при хранении |
Мелассо- |
I. Сухие вещества, % II. рН III. Сахар по поляриметру, % IV. Доброкачественность, % |
Один раз в квартал |
Показатели качества определяются в мелассе из каждого мелассохранилища |
|||||||||
|
1. Инвертный сахар, % |
|||||||||||||
|
2. Общее количество микроорганизмов в 1 г мелассы и их качественный состав |
|||||||||||||
|
Меласса при передаче в производство |
Мелассо- |
I. Сахар по поляриметру, % II. Инвертный сахар, % III. Раффиноза, % IV. Сумма сбраживаемых сахаров, % V. Азот общий и аминный, % VI. Нитриты по биопробе |
Перед началом переработки каждого нового хранилища |
Применяется методика определения количества калия при сухом озолении вещества |
|||||||||
|
VII. Калий (K 1. 3ола, % ("условная углекислая" и "за вычетом углекислого кальция") 2. Кальций (СаО), % 3. Магний (MgO), % 4. Цветность, мл 0,1 н. раствора йода на 100 мл 2%-ного раствора мелассы 5. Буферность, мл 1 н. раствора кислоты 6. Общее количество микроорганизмов в 1 г мелассы и их качественный состав 7. Сернистый ангидрид, % 8. Летучие кислоты, % |
|
Анализ проводится по мере надобности |
Содержание калия в мелассе можно определить при сухом озолении и ориентировочно по ускоренному методу |
||||||||||
|
Меласса, поступившая в производство |
Весы (или сборник производствен- |
I. Сухие вещества, % II. Сахар по поляриметру, % III. рН IV. Доброкачественность, % V. Плотность, г/см |
|
В каждой смене или 1 раз в сутки |
В средней пробе за смену или за 1 сут |
||||||||
|
VI. Сахар по поляриметру, % VII. Инвертный сахар, % VIII. Сумма сбраживаемых сахаров, % |
|
Через каждые 10 сут |
Показатели качества определяются в средней пробе, составляемой в течение 10 сут, в результате отбора мелассы в количестве 0,5 кг от каждого заполнения весов |
||||||||||
|
Основные материалы |
|||||||||||||
|
Аммоний сернокислый |
Мешки |
I. Мышьяк, % II. Сульфиды и сульфиты (по высоте черного кольца), мм 1. Азот, % 2. Влага, % |
При поступлении на завод партии |
Показатели качества определяют в пробе от поступившей партии |
|||||||||
|
Диаммоний- |
" |
I. Мышьяк, % II. Фтор, % |
То же |
То же |
|||||||||
|
1. Р 2. Азот, % 3. Влага, % |
|||||||||||||
|
Кислота ортофосфорная термическая (пищевая) |
Стеклянные бутыли |
I. Мышьяк, % Н |
При поступлении на завод партии |
Показатели качества определяются в пробе от посту- |
|||||||||
|
Аммиак водный |
Цистерны. Аммиаковозы. Железные бочки |
1. Содержание аммиака, % |
То же |
То же |
|||||||||
|
Экстракт кукурузный сгущенный |
Цистерны |
1. Сухие вещества, % 2. Кислотность, % молочной кислоты |
" |
" |
|||||||||
|
Хлористый калий |
1. Влага, % 2. Содержание хлористого калия, % |
" |
" |
||||||||||
|
Вспомогательные материалы |
|||||||||||||
|
Кислота серная техническая (контактная улучшенная марок А и Б или аккумуляторная) |
Цистерны. Контейнеры. Бочки. Стеклянные бутыли |
I. Мышьяк, % II. Плотность, г/см III. Моногидрат, % |
" |
" |
|||||||||
|
Магний сернокислый |
1. Влага, % 2. Содержание магния сернокислого, % |
" |
" |
||||||||||
|
Кислота олеиновая техническая марок А и Б |
Бочки. Стальные барабаны. Цистерны |
1. Температура застывания, °С 2. Влага, % |
" |
" |
|||||||||
|
Кислота молочная пищевая |
Бутыли. Деревянные парафиниро- |
1. Концентрация (сумма прямо титруемой и прямо нетитруемой кислоты молочной), % |
" |
" |
|||||||||
|
Известь хлорная |
Бочки. Барабаны |
1. Активный хлор |
При поступлении на завод партии и при ее хранении |
Показатели качества определяются в пробе от партии |
|||||||||
|
Подготовительное отделение |
|||||||||||||
|
Меласса разбавленная перед осветлением |
Емкость для разбавленной мелассы |
I. Сухие вещества, % II. рН III. Содержание мелассы 46%-ной сахаристости, кг/м |
Анализ проводится по мере надобности |
Плевако Е.А. Технология дрожжей. - М.: "Пищевая промышленность", 1970 |
|||||||||
|
Приточная меласса |
Емкость для осветленной мелассы |
1. Сухие вещества, % 2. Нитриты по биопробе 3. Температура нагрева мелассы при горячем способе осветления, °С Температура мелассы при подаче в дрожжерастильный аппарат (при горячем и холодном способах осветления), °С |
То же |
||||||||||
|
Раствор аммония сернокислого |
Сборник- |
1. Сухие вещества, % по сахарометру 2. рН |
После каждого заполнения сборника-соле- |
Табл.5, с. [3] |
|||||||||
|
Раствор диаммоний- |
То же |
1. Сухие вещества, % по сахарометру или рефрактометру 2. рН |
То же |
То же |
|||||||||
|
Раствор экстракта кукурузы |
Сборник |
1. Количество сухих веществ, % |
После заполнения сборника |
" |
|||||||||
|
Аммиак водный |
1. Аммиак, % |
По мере надобности |
|||||||||||
|
Отделение чистой культуры (ЧК) и естественно-чистой культуры (ЕЧК) |
|||||||||||||
|
Питательная среда в цехе чистой культуры |
Из аппаратов чистой культуры |
I. Температура нагрева мелассы и ее температура при подаче в дрожжерастильный аппарат, °С II. Сухие вещества, % III. рН IV. Нитриты |
Каждая стадия |
По мере надобности высевают на чашки Петри |
|||||||||
|
V. Посторонние микроорганизмы (посев) |
По мере надобности |
Высевают на чашки Петри |
|||||||||||
|
Кислотная обработка |
I. Кислотность, мл 0,1 н. раствора щелочи на 10 мл взвеси |
Каждый раз "кислотная обработка" |
|||||||||||
|
II. Продолжительность, мин |
То же |
||||||||||||
|
Культуральная среда (по бесприточным стадиям) |
Аппараты для выращивания чистой и естествен- |
I. Температура, °С II. Сухие вещества, % III. рН IV. Формольное число, мл 0,1 н. щелочи на 10 мл среды V. Почкующиеся клетки, посторонние дрожжи, бактерии (отсутствие, мало, много) |
|
Каждая стадия: в начале, через каждые 1-2 ч и в конце |
|||||||||
|
VI. Посторонние микроорганизмы (посев) |
По мере надобности |
||||||||||||
|
VII. Нитриты (отсутствие, мало, много) |
То же |
||||||||||||
|
VIII. Концентрация дрожжей, г/л |
Каждая стадия в начале, через 1-2 ч и в конце |
Определяют по водомерному стеклу, манометру и др. |
|||||||||||
|
IX. Набор среды в аппарате, м |
Каждая стадия в конце выращивания |
||||||||||||
|
X. Биомасса, кг |
|||||||||||||
|
XI. Температура воздуха, °С и его количество (в м |
Каждая стадия |
||||||||||||
|
Культуральная среда (по воздушно- |
Дрожже- |
I. Температура, °С II. Сухие вещества, % III. рН IV. Формольное число, мл 0,1 н. щелочи на 10 мл среды V. Концентрация дрожжей, г/л VI. Число, %, почкующихся клеток посторонних дрожжей бактерий (отсутствует, мало, много) |
|
В начале и конце и через 1-2 ч в течение всего процесса |
|||||||||
|
VII. Посторонние микроорганизмы (посев) |
По мере надобности |
||||||||||||
|
VIII. Нитриты (отсутствие, мало, много) |
1-2 раза в течение смены |
||||||||||||
|
IX. Набор среды в аппарате, м |
При складке, в конце выращивания и в течение процесса по мере надобности |
||||||||||||
|
X. Биомасса, кг |
По стадии в конце процесса |
||||||||||||
|
XI. Температура воздуха (в °С) и его количество (в м |
Каждая стадия |
||||||||||||
|
Бражка и промывная вода |
На выходе из сепаратора |
I. Количество дрожжевых клеток в бражке и промывной воде II. Окисляемость промывной воды |
Непрерывное наблюдение в процессе сепарирования |
||||||||||
|
Фильтрат из вакуумных фильтров или фильтр-прессов Дрожжи чистой культуры (ЧК) и естествен- |
Вакуум-фильтр или фильтр-пресс |
I. Количество дрожжевых клеток в фильтрате (в поле зрения микроскопа) II. Стойкость при 35 °С, ч III. Число, % посторонних дрожжей мертвых клеток бактерий (отсутствие, мало, много) агглютинация IV. Посторонние микроорганизмы (посев) |
|
1-2 раза в смену в процессе прессования |
Стойкость дрожжей определяется в случае прессования |
||||||||
|
5. Влажность, % 6. Кислотность, мг уксусной кислоты на 100 г дрожжей |
|
По мере надобности |
|||||||||||
|
7. Азот, % 8. Р 9. Мальтазная активность, мин |
|
То же |
|||||||||||
|
10. Температура, С 11. Концентрация дрожжей, г/л 12. Биомасса, кг |
|
После каждого заполнения сборника |
|||||||||||
|
Определение выращивания товарных дрожжей |
|||||||||||||
|
Культуральная среда |
Дрожже- |
I. Температура, °С II. Сухие вещества, % III. рН IV. Формольное число, мл 0,1 н. щелочи на 10 мл среды |
|
При складке и через 1-2 ч в течение всего процесса выращивания |
|||||||||
|
V. Концентрация дрожжей, г/л |
В начале и конце и по ходу процесса через 1-2 ч |
||||||||||||
|
VI. Нитриты (отсутствие, мало, много) VII. Число, % почкующихся клеток посторонних дрожжей мертвых клеток бактерий (отсутствие, мало, много) |
|
1-2 раза в течение смены |
|||||||||||
|
VIII. Набор в аппарате, м |
При складке, в конце выращивания и в течение процесса по мере надобности |
Определяют по водомерному стеклу, манометру и др. |
|||||||||||
|
IX. Биомасса, кг |
В конце процесса |
||||||||||||
|
X. Температура воздуха (в °С) и его количество (в м XI. Спирт в бражке, % |
|
По мере надобности |
|||||||||||
|
Культуральная среда |
Отборочный аппарат |
I. pH II. Накопление дрожжей, г/л III. Температура, °С |
|
Через каждые 2 ч |
|||||||||
|
IV. Формольное число, мл 0,1 н. щелочи на 10 мл среды |
Перед сепарированием |
||||||||||||
|
V. Набор среды в аппарате, м |
Ежечасно |
По водомерному стеклу, манометру и др. |
|||||||||||
|
Выделение товарных дрожжей |
|||||||||||||
|
а) сепарирование |
|||||||||||||
|
Бражка и промывная вода |
На выходе из сепаратора |
I. Количество дрожжевых клеток в бражке и промывной воде (в поле зрения микроскопа) |
Непрерывное наблюдение в процессе сепарирования |
||||||||||
|
II. Окисляемость промывной воды |
В конце сепарирования |
||||||||||||
|
Молоко дрожжевое |
Сборник |
I. Температура, °С |
После каждого заполнения сборника |
||||||||||
|
II. Концентрация дрожжей, г/л |
|||||||||||||
|
б) прессование |
|||||||||||||
|
Фильтрат из вакуум-фильтров или фильтр- |
На выходе из вакуум-фильтра или фильтр-пресса |
I. Количество дрожжевых клеток в фильтрате (в поле зрения микроскопа) |
1-2 раза в смену в процессе прессования |
||||||||||
|
в) готовая продукция прессованные дрожжи |
|||||||||||||
|
Прессованные дрожжи |
Формовочно- |
I. Органолептические показатели |
Каждая партия |
Для анализов отбирают среднюю пробу дрожжей в начале, середине и конце формования. Допускается отбор пробы и только в начале формования |
|||||||||
|
II. Влажность, % |
То же |
||||||||||||
|
III. Подъемная сила, мин |
" |
||||||||||||
|
IV. Кислотность, мг уксусной кислоты на 100 г дрожжей, в день выработки и после 12 сут хранения |
" |
Кислотность дрожжей после 12 сут хранения определяется по мере надобности |
|||||||||||
|
V. Стойкость при 35 °С, ч |
" |
Для определения стойкости отбирается брусок |
|||||||||||
|
VI. Стойкость дрожжей при температуре хранения от 0 до 4 °С, сут |
" |
||||||||||||
|
VII. Масса бруска, г |
По мере надобности |
Контролируется упаковка и маркировка |
|||||||||||
|
VIII. Азот, % |
То же |
||||||||||||
|
IX. Р |
" |
||||||||||||
|
X. Число, % посторонних дрожжей мертвых клеток бактерий (мало, много) |
" |
||||||||||||
|
XI. Посторонние микроорганизмы (посев) |
По мере надобности |
Посев на чашки Петри |
|||||||||||
|
XII. Мальтазная активность, мин |
То же |
||||||||||||
|
г) холодильная камера |
|||||||||||||
|
Холодильная камера |
I. Температура, °С |
1 раз в смену |
Определяется ртутным термометром, лагометром ЛР-64 |
||||||||||
|
II. Относительная влажность воздуха, % |
То же |
Меклер В.Я., Овчинников П.А. Промышленная вентиляция и кондициониро- |
|||||||||||
|
Отделение сушки |
|||||||||||||
|
Прессованные дрожжи, поступающие на сушку |
Вакуум-фильтр или фильтр-пресс |
I. Влажность, % II. Подъемная сила, мин |
|
В партии дрожжей, поступающей на сушку |
|||||||||
|
III. Посторонние дрожжи, % Бактерии (мало, много) IV. Азот, % |
|
По мере надобности |
|||||||||||
|
Воздух, поступающий в сушилку и отходящий из нее |
При входе и выходе из сушилки |
I. Температура поступающего и отходящего воздуха, °С |
2-3 раза в смену |
Семихатова Н.М., Чулина Е.П. и др. Технология производства сушеных дрожжей. М.: Пищевая промышленность, 1976 |
|||||||||
|
Дрожжи из сушилки |
Сушилка |
1. Влажность дрожжей, % |
По мере надобности |
||||||||||
|
Сушеные дрожжи (готовая продукция) |
I. Органолептические показатели |
В партии сушеных дрожжей |
|||||||||||
|
II. Влажность, % |
То же |
||||||||||||
|
III. Подъемная сила, мин |
" |
||||||||||||
|
IV. Длительность хранения, мес |
Экспресс-метод описан в книге Семихатовой Н.М. и др. Технология производства сушеных дрожжей. М.: Пищевая промышленность, 1976. |
||||||||||||
|
V. Масса единичной фасованной и нефасованной продукции |
1 раз в смену |
||||||||||||
|
VI. Количество мертвых клеток, % |
По мере надобности |
||||||||||||
|
Склад для сушеных дрожжей |
|||||||||||||
|
Склад для сушеных дрожжей |
I. Температура, °С |
1 раз в сутки |
Определяется ртутным термометром, лагометром ЛП-64 |
||||||||||
|
II. Относительная влажность воздуха, % |
То же |
Меклер В.Я., Овчинников П.А. Промышленная вентиляция и кондициониро- |
|||||||||||
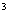
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Инструкция по технохимическому контролю пивоваренного производства. - М.: Пищевая промышленность, 1975. - 214 с.
2. Контроль производства хлебопекарных дрожжей / [О.А.Бакушинская, Л.Д.Белова, В.И.Буканова и др.]. - М.: Пищевая промышленность, 1978. - 168 с.
3. Плевако Е.А., Бакушинская О.А. Микробиологический и химико-технологический контроль дрожжевого производства. - М.: Пищевая промышленность, 1964. - 269 с.